 |


Обмен фенилаланина и тирозина
|
|
|
|
Фенилаланин – незаменимая аминокислота, т.к. в клетках животных не синтезируется бензольное кольцо.
Основная масса фенилаланина утилизируется двумя путями – превращается в тирозин (90%) или включается в состав белков. Обмен тирозина значительно сложнее: тирозин используется для синтеза белков, служит предшественником катехоламинов, меланина, тироксина, а также может подвергаться катаболизму до СО2 и Н2О.
Катаболизм фенилаланина и тирозина. В результате ряда ферментативных превращений эти аминокислоты превращаются в фумарат и ацетоацетат.
Превращение фенилаланина в тирозин катализируется ферментом фенилаланинмонооксигеназой, коферментом которой служит тетрагидробиоптерин (ТГБП). Для регенерации последнего используется НАДФН·Н+. Превращение фенилаланина в тирозин скорее нужно для удаления избытка фенилаланина, чем для образования тирозина, поскольку недостатка в тирозине обычно не бывает. При врожденном отсутствии этого фермента развивается заболевание фенилкетонурия.
Фенилкетонурия характеризуется нарушением обмена фенилаланина, в результате последний не может превращаться в тирозин и поэтому накапливается во всех жидкостях организма. Некоторые превращения фенилаланина, количественно несущественные у здорового человека, становятся заметными при фенилкетонурии. Наиболее значительным из них является переаминирование фенилаланина с образованием фенилпирувата. В основе самого названия болезни лежит высокое содержание этого фенилкетона в моче. Из фенилпирувата далее могут образовываться фениллактат, фенилацетат и О-гидроксифенилацетат.
Phe → фенилпируват → фенилацетат → фенилацетилглутамин
|
|
|
Конъюгат фенилацетата с глутамином выводится из организма с мочой.

Различают 2 формы фенилкетонурии: 1) классическая – наследственное заболевание связано с мутацией в гене фенилаланинмонооксигеназы. Наиболее тяжелые проявления – нарушение умственного и физического развития, судорожный синдром. 2) вариантная - следствие мутаций в генах, контролирующих метаболизм тетрагидробиоптерина. При этой форме клинические проявления близки, но не во всем совпадают с классической формой.
Нарушение умственного и физического развития при фенилкетонурии связано с токсическим действием на клетки мозга высоких концентраций фенилаланина, фенилпирувата и фениллактата. Большие концентрации Phe ограничивают транспорт Tyr и Trp через гематоэнцефалический барьер и тормозят синтез нейромедиаторов.
Тетрагидробиоптерин необходим для реакций гидроксилирования не только Phe, но и Tyr, и Trp. Поэтому при недостатке этого кофермента нарушается метаболизм всех 3-х аминокислот, в том числе синтез нейромедиаторов – катехоламинов и серотонина. Заболевание характеризуется тяжелыми неврологическими нарушениями и ранней смертью (злокачественная фенилкетонурия).
При фенилкетонурии имеют место и другие нарушения аминокислотного обмена. Так, кожа и волосы у больных фенилкетонурией светлее, чем у их сибсов. Это обусловлено ингибированием реакции гидроксилирования тирозина – первого этапа в образовании пигмента меланина под влиянием высокой концентрации фенилаланина.
Лечение фенилкетонурии сводится к приему пищи с низким содержанием фенилаланина. Задача состоит в том, чтобы поступление фенилаланина в организм при данном заболевании не превышало потребности в нем для роста и замещения.
В связи с резко выраженной умственной отсталостью, развивающейся при фенилкетонурии, важное значение приобретает ранняя диагностика. С этой целью исследуют мочу новорожденного, добавляя в нее FeCI3. В присутствии фенилпирувата развивается оливково-зеленое окрашивание. Еще более надежным тестом считается определение фенилаланина в крови.
|
|
|
Частота встречаемости фенилкетонурии составляет 1 случай на 20000 новорожденных. Болезнь наследуется как аутосомный рецессивный признак. Гетерозиготы, составляющие ~ 1,5% популяции, не обнаруживают видимых отклонений от нормы. Однако гетерозиготных носителей гена фенилкетонурии можно обнаружить с помощью теста толерантности к фенилаланину или по измерению кинетики исчезновения внутривенно введенного фенилаланина. Эти тесты используются в генетической консультации для определения риска рождения больного ребенка.
Другим наследственным заболеванием, развивающимся как результат нарушения метаболизма тирозина, является алкаптонурия. Его непосредственной причиной является дефект фермента гомогентизат-диоксигеназы. В результате гомогентизат накапливается в жидкостях организма и выделяется с мочой, которая при стоянии чернеет, поскольку гомогентизат окислятся и полимеризуется в меланиноподобное соединение. Болезнь обычно обнаруживают по появлению черных пятен на пеленках. Других клинических проявлений и, прежде всего, нарушения умственного развития при данном заболевании не наблюдается.
Биосинтез меланина
В пигментных клетках из тирозина образуется пигмент меланин (от греч. melas – черный). При действии тирозинмонооксигеназы тирозин окислятся в дигидроксифенилаланин (ДОФА). ДОФА под действием тирозиназы – ключевого фермента всего процесса биосинтеза меланина превращается в ДОФА-хинон, из которого в результате неферментативных реакций образуется меланин. Меланин представляет собой группу полимерных соединений с неупорядоченной структурой. Цвет кожи и глаз зависит от количества и распределения меланоцитов и содержания в них меланина. Врожденное отсутствие тирозиназы в меланоцитах или отсутствие самих меланоцитов проявляется как альбинизм. Для этого заболевания характерны отсутствие пигментации кожи, волос и радужной оболочки глаз, сниженная острота зрения.
Обмен гистидина
Катаболизм гистидина происходит путем его внутримолекулярного дезаминирования с образованием уроканиновой кислоты, которая затем через ряд реакций превращается в аммиак, одноуглеродный фрагмент, соединенный с тетрагидрофолатом, и глутаминовую кислоту. Дезаминирование гистидина катализируется гистидин-аммиаклиазой, которая содержится в печени и коже:
|
|
|

Уроканиновая кислота превращается в имидазолонпропионовую кислоту под действием уроканиназы, которая содержится только в печени:
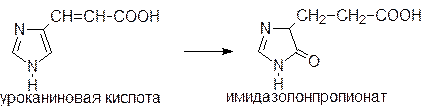
И гистидин-аммиаклиазой, и уроканиназа появляются в крови при заболеваниях печени и измерение их активности используется для диагностики. Известна наследственная болезнь - гистидинемия, связанная с дефектом гистидин-аммиаклиазой. Для этого заболевания характерно повышенное содержание гистидина в тканях и нарушение физического и умственного развития.
|
|
|
