 |


Получение ДНК-фрагментов желаемого размера
|
|
|
|
ДНК, подвергаемая частичному гидролизу, должна быть достаточно высокомолекулярной. Методика приготовления образцов ДНК та же, что и для пульс-электрофореза, с той лишь разницей, что количество ДНК рассчитывается для препаративных целей.
1. Исходя из того, что одна клетка млекопитающего содержит 6,7 пкг ДНК, необходимо вырастить их столько, чтобы получить достаточное количество ДНК. Обычно для создания библиотеки требуется 200 мкг ДНК, что соответствует приблизительно 3х107 клеткам. ДНК должна быть очень высокого качества, поэтому очень важно растить клетки в оптимальных условиях. Можно приготовить клетки и из периферической крови центрифугированием в смеси Ficoll-Hypaque.
2. После сбора клеток аккуратно просчитайте их количество в гемоцитометре и суспендируйте в таком объеме фосфатно-солевого буфера, чтобы концентрация клеток составляла примерно 2хЮ7/мл. Затем суспензию быстро смешайте с равным объемом расплавленной 2%-ной низкоплавкой агарозы в 125 мМ ЭДТА при 40°С. Залейте в форму. Кроме стандартных ячеек форма должна иметь ячейки для блоков размером 2x8x135 мм, которые служат для получения ДНК в препаративных количествах. Полезно приготовить 10–20 стандартных блоков с ДНК для тест-гидролизатов и два больших блока непосредственно для опытов.
3. Обработайте ДНК протеиназой К в присутствии больших количеств ЭДТА и N-лаурилсаркозина, как описано ранее.
4. Очищенную ДНК проверьте на нативность и на присутствие нуклеаз. Для этого инкубируйте половину содержимого блока при 37°С 3 ч с 10 мм MgCl2 и нанесите эту смесь, а также необработанную половину на OFAGE-гель или на гель для электрофореза в инвертированном поле. Определите размер ДНК. Необработанная ДНК должна практически полностью остаться в лунке, а в обработанном магнием препарате не должно быть низкомолекулярных примесей. Только в этом случае можно использовать препараты для «прыжков по хромосоме». Если же в препаратах, обработанных магнием, налицо признаки деградации, значит они загрязнены нуклеазами и надо повторить обработку протеиназой К. Как правило, этого бывает достаточно, чтобы избавиться от загрязнений.
|
|
|
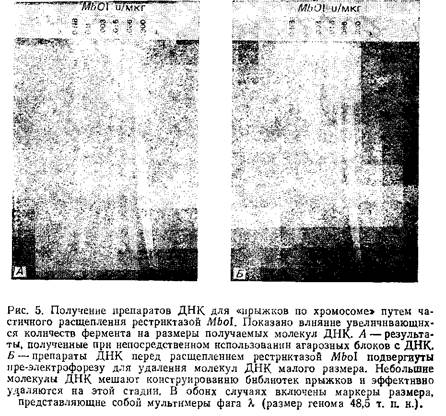
5. Из некоторых источников ДНК постоянно выделяется с небольшим количеством низкомолекулярных примесей размером 50–100 т. п.н., видимо из мертвых клеток. Наиболее характерно это для лимфобластов. Такие деградировавшие молекулы ДНК могут сильно искажать результаты «прыжков по хромосоме», поэтому необходимо удалить их из агарозных блоков перед обработкой рестриктазами. Для этого блоки помещают в лунки OFAGE-геля и проводят пульс-электрофорез в течение 2–3 ч с интервалами между импульсами 20 с. Молекулы ДНК размером менее 100 т. п.н. выходят из блоков в гель, а высокомолекулярная ДНК остается практически без изменения. Блоки затем можно изъять из геля, получив, таким образом, высококачественный материал для дальнейшего исследования.
6. Проведите контрольную рестрикцию половинок агарозных блоков различными концентрациями Mbol. Согласно данной методике, в каждой половине блока содержится примерно 3,3 мкг ДНК, следовательно, концентрация фермента составит от 0,01 ед./мкг до 0,045 ед./мкг. Остановите реакцию добавлением 10 мкл ЭДТА. Обработанные рестриктазой образцы поместите в гель и проведите пульс-электрофорез. Это поможет определить концентрацию фермента, оптимальную для получения фрагментов нужного размера. На рисунке наглядно продемонстрирована необходимость пре-электрофореза для удаления низкомолекулярных примесей ДНК перед обработкой рестриктазами.
7. При использовании больших агарозных блоков пропорционально увеличьте количество фермента, но так, чтобы концентрации фермента в ед./мкл и ДНК в мкг/мл остались прежними.
|
|
|
8. Обработанные рестриктазой препаративные блоки поместите в гель и, используя соответствующие маркеры, проведите пульс-электрофорез с интервалами между импульсами, позволяющими хорошо отделить фрагменты нужной величины.
Если используется OFAGE-гель, маркеры следует наносить в середине и по обоим краям геля. Мы обнаружили, что для получения прямых полос молекул ДНК желаемых размеров лучше использовать CHEF-гели и гель-электрофорез в инвертированном поле.
9. Важно, чтобы ДНК, которую предстоит клонировать, не подвергалась ультрафиолетовому облучению. Поэтому отрежьте края геля, содержащие маркеры, а также небольшое количество рестрицированной геномной ДНК, окрасьте их бромидом этидия и визуализируйте в ультрафиолете. По полученным данным определите участок геля с интересующей вас ДНК.
10. Вырежьте нужный участок геля. На этой и последующих стадиях важно использовать безнуклеазные реактивы и инструменты. Выделять ДНК из геля для получения кольцевой молекулы можно двумя способами. Нам представлялось удобным делать это методом электроэлюции. Поместите кусочек геля с ДНК в диализный мешок, залейте четырьмя объемами электрофорезного буфера, а затем поместите мешок в камеру для горизонтального электрофореза. Элюируйте ДНК из геля в течение двух часов 0,5хТВЕ-буфером при постоянном напряжении 100 В. Чтобы убедиться, что вся ДНК элюирована, можно вскрыть диализный мешок, достать гель и окрасить бромидом этидия. После удаления геля снова запечатайте диализный мешок и отдиали-зуйте ДНК против 10 мм Трис, рН 7,4, 1 мм ЭДТА с несколькими сменами буфера, подготовив таким образом препарат для лигирования.
11. Препаративные гели для определения размеров ДНК можно готовить из легкоплавкой агарозы. И тогда выделять ДНК из них можно, вырезая нужные участки и расплавляя их при 65°С. После того как гель расплавится, ДНК доводят до нужной концентрации и проводят лигирование в присутствии агарозы, которая не ингибирует эту реакцию. В настоящее время нет оснований предпочитать один метод другому. Важно лишь избегать операций, которые могли бы нарушить целостность молекул ДНК после извлечения их из геля. Нельзя центрифугировать, встряхивать, пипетировать препараты. И желательно как можно скорее приступать к их лигированию.
|
|
|
Циклизация
Успех или неудача «прыжков по хромосоме» в решающей степени определяются возможностью лигировать большие сегменты ДНК с образованием кольцевых молекул. Анализ стратегии этого метода, схематически изображенной на рис. 4, показывает, что тандемное лигирование молекул ДНК дает сцепленные фрагменты, соединяющие в себе две случайные последовательности. Такое сцепление приводит к образованию аномальных клонов, которые провоцируют «прыжки по хромосоме» из данной стартовой точки на какую-то случайную последовательность генома. Свести к минимуму риск возникновения такого события можно, проводя лигирование при достаточно низких концентрациях ДНК так, чтобы на долю тандемных лигирований приходилось менее 5–10%.
Теория лигирования ДНК при низких концентрациях была разработана в 50-е годы Джекобсоном и Стокмайером. Для некоторых размеров последовательностей ДНК теоретические прогнозы подтвердились экспериментально. Очень полезно, например, следующее соотношение:

где j – концентрация молекул ДНК контурной длины / и сегментной длины b, при которой с равной вероятностью лигируются как разные молекулы, так и концы одной молекулы. Очевидно, что для эффективной циклизации лучше работать с концентрациями гораздо более низкими, чем /. Подставляя в уравнение известные параметры ДНК для водного раствора, его можно преобразовать следующим образом:

где т.п.н. – длина ДНК в тысячах пар нуклеотидов. При данной концентрации ДНК i – доля лигирований, приводящих к циклизации. Чтобы эта доля достигала 90%, работать следует с концентрацией ДНК, определяемой уравнением:
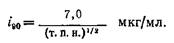
Обычно для получения 3х106 клонов в библиотеке, необходимо после расщепления циклизованных молекул ДНК иметь 0,5 мкг сцепленных фрагментов. Средний размер такого фрагмента 5 т. п.н., а это значит, что для осуществления прыжков в 100 т. п.н. необходимо иметь 10 мкг фракционированной по размеру ДНК; для прыжков в 200 т. п.н. – уже 20 мкг. На интенсивность лигирования влияют два фактора: 1) количество ДНК, необходимое для получения достаточного числа сцепленных фрагментов и создания геномной библиотеки. Это количество возрастает линейно с увеличением размера прыжка; 2) необходимость преимущественного получения циклизованных молекул. Интенсивность лигирования с циклизацией возрастает пропорционально квадратному корню длины молекул ДНК. Поэтому с увеличением размера прыжка объем лигазной смеси должен возрастать пропорционально 3/2 степени длины молекулы ДНК. В табл. 1 приведены стандартные параметры для создания геномной библиотеки, рассчитанной на прыжки в 100 т. п.н.
|
|
|
Очень важно маркировать в кольцевых молекулах места соединения фрагментов с тем, чтобы селективно клонировать их на последующих стадиях. В качестве селективного маркера мы использовали ген супрессорной тРНК – supF, хотя возможны и другие способы селекции. Супрессорный ген должен иметь концы, комплементарные Mfeol-концевым фрагментам геномной ДНК – Для этой цели было взято несколько supF с различающимися концевыми последовательностями, полученными при обработке рестриктазной Ват.
Ниже представлена методика реакции циклизации.
1. Используя приведенные выше уравнения, растворите ДНК и доведите раствор до нужной концентрации в 50 мМ Трис, рН 7,4, 1 мМЭДТА.
2. Добавьте 100 или 500-кратный молярный избыток ДНК BamHI-фрагмёнтов supF, предварительно проверенных на эффективность лигирования. Для этого проведите самолигирование и последующий анализ геля. По нашему мнению, наиболее удобно получать фрагменты генов supF электрофорезом с последующей электроэлюцией. Важно, чтобы фрагмент был максимально очищен от плазмиды, поскольку даже незначительные количества ее могут проявиться в окончательной библиотеке в виде клонов, гибридизующихся с зондом, несущим плазмиду. Оставьте смесь supF ДНК с геномной ДНК на полчаса.
3. Доведите концентрацию магния до 10 мМ и оставьте смесь еще на 10 мин, чтобы установилось равновесие. Затем добавьте ДНК-лигазу бактериофага Т4 до конечной концентрации 1–2 ед./мкл. Лигируйте 12 ч при 14°С, затем добавьте вторую порцию лигазы и лигируйте еще 12 ч. Осадите циклизовавшуюся ДНК этанолом, добавив 20 мкг дрожжевой тРНК-носителя. Отцентрифугируйте осадок при 23000 об/мин в роторе SW27. Ресуспендируйте осадок в 100 мкл ТЕ и инактивируйте все нелигировавшиеся концы, либо обработав их щелочной фосфатазой, либо добавив фрагмент Клёнова ДНК-полимеразы I. Эта стадия очень важна, так как, если лигирование при низких концентрациях происходит не полностью, свободные фрагменты могут аномально лигироваться с вектором. Экстрагируйте ДНК фенолом, осадите и, растворив, снова обработайте EcoRl.
|
|
|
5. Для контроля очень полезно на каждой стадии отбирать небольшие аликвоты и подвергать их электрофорезу в 1,4%-ном агарозном геле с последующим переносом на нитроцеллюлозу или полиамид и блот-гибридизацией с supF-гтом. В случае успешной реакции циклического лигирования ДНК гена supF будет давать полосы в виде лестницы, и небольшое ее количество должно оказаться в той зоне геля, которая соответствует высокомолекулярной ДНК. После обработки рестриктазой ЕсоЩ лесенка supF-гена должна остаться, а полосы из высокомолекулярной области должны расщепиться на множество фрагментов размером от 1 до 20 т. п.н.

6. ДНК, обработанную ЕсоШ, экстрагируйте фенолом и осадите этанолом.
Клонирование и скрининг
На этой стадии обработанные кольцевые молекулы геномной ДНК уже можно лигировать и производить селекцию сцепленных фрагментов. Для селекции геномные фрагменты встраивают в фаговый вектор, несущий амбермутации по крайней мере двух генов белковой оболочки, упаковывают фаговую ДНК in vitro и инфицируют клетки бактерии-хозяина, лишенные функции supF. Формировать бляшки на таком газоне могут только те фаговые частицы, геномы которых содержат собственные supF-гены.
Теоретически подходит любой фаговый вектор, несущий амбер-мутации и клонирующий сайт. Желательно, конечно, чтобы этот вектор обеспечивал максимальную клонирующую емкость. Идеальной можно считать ситуацию, когда кольцевые геномные фрагменты расщеплялись бы лишь частично мелкощепящим ферментом, а затем лигировались в вектор большой емкости, чтобы не было дискриминации фрагментов, обусловленной расположением сайтов рестрикции. Однако обязательные потери, происходящие при частичном расщеплении, оказываются препятствием на пути создания полной библиотеки. Имеющиеся у нас в настоящее время стандартные геномные библиотеки для «прыжков по хромосоме» были получены в результате полного расщепления кольцевых геномных фрагментов рестриктазой EcoRl. Можно использовать и другие ферменты, для которых имеются амбермутантные фаговые клонирующие векторы.

Анализ клонов
После очистки бляшек можно приготовить минилизат ДНК из клонов, руководствуясь любой стандартной методикой. Использование на этой стадии в качестве газона LE392 позволяет получить несколько больший выход фаговой ДНК, чем при посеве на МС1061. Обработка рестриктазой ЈcoRI позволит определить размер инсерционного фрагмента. Если в данном клоне имеется более одного fcoRI-фрагмента, это значит, что при клонировании в один вектор были лигированы две вставки. В таком случае следует провести блот-гибридизацию данного клона с исходным зондом и с supF, чтобы определить, находятся ли эти вставки в одном EcoRI-фрагменте или в разных. В первом случае с клоном можно продолжать работать дальше традиционными методами; во втором случае он не представляет ценности для дальнейших исследований.
ZTcoRI-фрагмент полезно переклонировать в плазмиду, чтобы упростить работу по его изучению. Субклоны можно легко идентифицировать, благодаря наличию SHpF-маркера. Рестрицированный минилизат ДНК можно лигировать в pBR322, предварительно обработанную ЈcoRI и фосфатазой, и полученной рекомбинантной ДНК трансформировать штамм бактерии-хозяина, несущий какую-либо амбермутацию в lacZ, например CARD-15. Высев культуры на агар Мак-Конки с ампициллином позволяет практически сразу выявить интересующие колонии, так как они способны сбраживать лактозу и окрашены в пурпурный цвет в отличие от остальных, шмеющих розовую окраску.
Для того чтобы разделить стартовую и конечную точки прыжка, очень полезно использовать присутствие сайта в центре гена supF. Особенно эффективно это в том случае, когда имеющийся в pBR322 сайт Aval элиминирован. Мы добивались этого, проводя последовательно обработку плазмиды рестриктазой Aval, достраивание фрагментом Клёнова и повторное лигирование. Отделить половины клонированного фрагмента можно, обрабатывая субклоны EcoRl, Aval и обеими рестриктазами одновременно. Лиасайты встречаются иногда в геномных последовательностях, что несколько затрудняет составление карты. Однако при помощи блот-гибридизации рестрицированных субклонов с исходным зондом, supF и геномной ДНК человека, позволяющей локализовать повторы, обычно легко удается построить рестрикционную карту и идентифицировать уникальные последовательности. А это, в свою очередь, позволяет определить, является ли данный прыжок эффективным. Результаты блот-гибридизации представляют особый интерес, так как с их помощью удается идентифицировать ближайшие к супрессорному гену фрагменты.

В случае же использования supF с дополнительными вставками редко встречающихся сайтов рестрикции разделение половинок фрагмента, составляющего прыжок, упрощается, и зонды можно получать непосредственно из минилизатафаговой ДНК. После того как установлено, что фрагмент – «прыжок» представлен в геноме единичной копией, надо провести его гибридизацию с блотом геномной ДНК из гибридных соматических клеток, чтобы убедиться, что прыжок осуществлен в пределах нужной хромосомы. Это очень важный момент, так как при создании геномной библиотеки всегда существует опасность нециклического лигирования.
|
|
|
12 |
