 |


Немецкий институт по стандартизации е.В. (ред.). DIN 28004, часть 1-4: Блок-схемы технологических установок (1988)
|
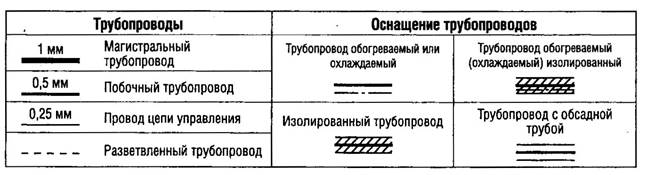
|
|
|


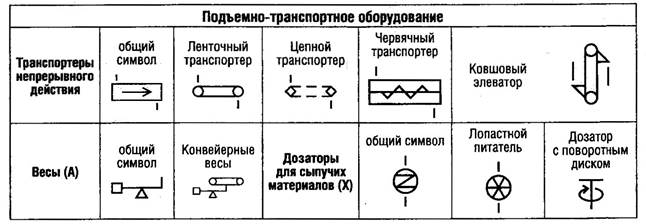
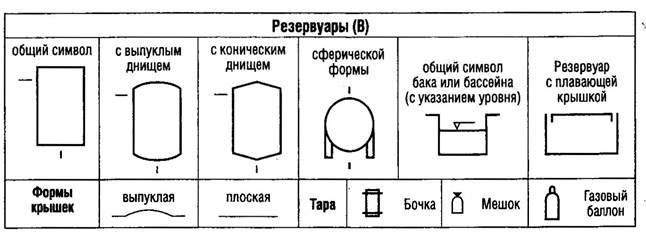

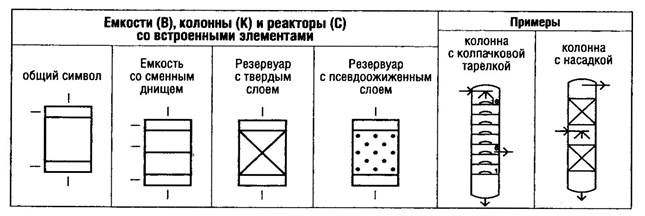
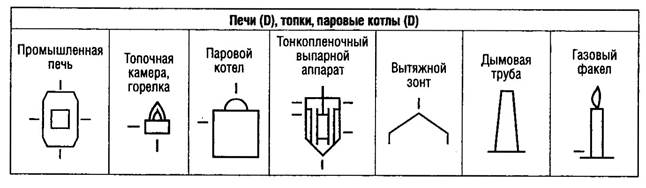



В мировой практике одним из важнейших документов, определяющим требования к производству и контролю качества лекарственных средств для человека и животных, являются "Правила производства лекарственных средств" - "Good Manufacturing Practice for Medicinal Products (GMP)".
Такая система включает фармацевтическое законодательство, порядок регистрации продуктов и лицензирования предприятий по производству, импорту, оптовой и розничной торговле, фармакопейную программу, лабораторную службу (независимые от производства контрольно-аналитические лаборатории, институты, центры), инспекторат, программу мониторинга неблагоприятных реакций лекарств.
Главная задача проектирования фармацевтических заводов – создание концепции производства фармацевтики, соответствующей регламентам GMP. Развитие современных производств сопровождается значительным усложнением технологических схем, созданием энерготехнологических циклов, машин и аппаратов сложных конструкций, работающих в условиях агрессивных сред, высоких температур и давлений. В связи с этим при проектировании необходимо решать проблемы охраны окружающей среды, применения новых материалов, обеспечения надежности технологического оборудования, безопасности жизнедеятельности обслуживающего персонала. Все это требует совершенствования самого процесса проектирования, повышения качества проектной документации, четкого определения совокупности нормативных документов по отдельным стадиям проекта.
На уровне производственного предприятия, интегрированного с научными исследованиями, центральным элементом системы обеспечения качества является GMP - "Good Manufacturing Practice for Medicinal Products" - требования к производству и контролю качества лекарственных средств для человека и животных - "Правила производства лекарственных средств".
|
|
|
Правила GMP устанавливают требования к системе управления качеством, контролю качества исходного сырья, персоналу, помещениям и оборудованию, документации, производству продукции и проведению анализов по контрактам, рекламациям, порядку отзыва продукции и организации самоинспекций.
В основе концепции GMP лежит принципиально новый подход к обеспечению качества лекарственных средств, а именно: переход от контроля качества готовой продукции к обеспечению ее качества во время процесса производства. При этом объектом контроля в первую очередь становятся сам процесс производства и различные производственные факторы (здания, помещения, оборудование, персонал и т.д.), т.е. работа по стандартам GMP полагает самоинспекцию: ежедневный внутренний контроль способа производства и качества выпускаемой продукции на основе соответствующих нормативов и приборной базы. Без постоянного контроля нельзя поддерживать производство в соответствии с стандартами GMP.
Стандарты GMP базируются на необходимости устранения негативных моментов в производственном процессе, в результате учета тех факторов, которые могут нанести ущерб готовой продукции. Причина постоянно возрастающих требований к обеспечению качества при разработке, исследованиях, производстве и распространении лекарственных средств обусловлена тем, что качество неразрывно связано с безопасностью и
эффективностью препаратов и, следовательно, со здоровьем и безопасностью
каждого отдельного пациента и общества в целом.
В РК Республиканским государственным предприятием «Национальный центр экспертизы лекарственных средств, изделий медицинского назначения и медицинской техники» Министерства здравоохранения Республики Казахстан разработан Госстандарт РК производство лекарственных средств. Надлежащая производственная практика. СТ РК 1617-2006. Утвержден и введен в действие приказом председателя комитета по техническому регулированию и метрологии Министерства индустрии и торговли Республики Казахстан №575. В настоящем стандарте учтены основные нормативные положения следующих международных документов: Правила производства лекарственных средств (GMP) Европейского Союза (GMP ЕС) 4 пересмотренное издание, Надлежащая производственная практика фармацевтических препаратов ВОЗ (официальный перевод на русский язык Good manufacturing practices for pharmaceutical products (Who Technical Report Series, №823, Проект для составления национальных руководств стран СНГ «Надлежащая производственная практика фармацевтических препаратов», Директива 2001/83/ЕС «Правила руководства по GMP о независимом контроле качества и Уполномоченном лице». В стандарте реализованы нормы законов Республики Казахстан «О лекарственных средствах», «О техническом регулировании», «О языках в Республике Казахстан», «О системе здравоохранения», «Об охране здоровья граждан».
|
|
|
Каждому из важнейших аспектов пригодности к применению лекарственных препаратов соответствует кодекс профессиональной деятельности. Правила GMP обеспечивают соблюдение фармацевтических аспектов качества, правила GCP позволяют объективно оценить, прежде всего, эффективность, правила GLP содействуют достижению безопасности препаратов.
Термины, применяемые при проектировании производств в соответствии с системой стандартов надлежащей производственной практики (GMP) Всемирной организации здравоохранения (ВОЗ):
Воздушный шлюз (airlock) - ограниченное пространство с двумя или несколькими дверьми между двумя или несколькими помещениями, например, разных классов чистоты, служащее для контроля потока воздуха между этими помещениями, когда в них необходимо войти. Воздушные шлюзы предназначаются и используются для перемещения как людей, так и товаров.
Чистая зона (clean area) - зона, в которой контролируется окружающая среда на контаминацию частицами и микроорганизмами, построенная и эксплуатируемая таким образом, чтобы уменьшить проникновение, образование и сохранение контаминантов внутри зоны.
|
|
|
Перед проектированием производства между Заказчиком и Исполнителем должен быть составлен контракт, в котором определены их обязанности по отношению друг к другу касательно производства и контроля продукции. Технические аспекты контракта должны составляться компетентными лицами, имеющими соответствующие знания фармацевтической технологии, аналитической химии и GMP. Целью самоинспекции является оценка соответствия
производителя требованиям GMP во всех аспектах производства и конт
роля качества.
Должны быть разработаны письменные инструкции по само
инспекции, чтобы обеспечить минимальные и единообразные типовые
требования. Эти инструкции могут включать в вопросники по требованиям GMP, охватывающие, по меньшей мере, следующие пункты:
(a) персонал;
(b) помещения, включая помещения для персонала;
(c) содержание зданий и обслуживание оборудования;
(d) хранение исходного сырья и готовой продукции;
(e) оборудование;
(f) технологический процесс и контроль в процессе производства;
(g) контроль качества;
(h) документация;
(i) санитария и гигиена;
(j) программы валидации и ревалидации;
(к) калибровка приборов или систем измерения;
(1) процедуры отзыва;
(т) организация рассмотрения жалоб;
(п) контроль этикеток;
(о) результаты предыдущих самоинспекций и любые предпринятые корректирующие действия.
Первые официальные требования GMP появились в США в 1963 году. К настоящему времени практически во всех странах, производящих лекарственные препараты, приняты либо национальные требования GMP (по данным ВОЗ более, чем в 30 странах), либо один из международных документов.
Требования GMP признаны международными организациями, деятельность которых связана с производством или распределением медикаментов, например ЮНИДО, Мировым банком и др. На основе рекомендаций ВОЗ и документов Европейского сообщества в рамках Конвенции были разработаны единые требования GMP, получившие широкую известность за пределами Европы.
|
|
|
Принципы GMP
Помещения и оборудование
Общие положения
Место расположения, проект, строительство, монтаж, оснащение и обслуживание помещений и оборудования должны соответствовать характеру выполняемых работ. Планировочные решения помещений и конструкция оборудования должны минимизировать риск ошибок, предусматривать проведение эффективной уборки и обслуживания с целью предотвращения перекрестного загрязнения, появления пыли или грязи и, в общем случае, устранения любого фактора, ухудшающего качество продукции.
Помещения
Общие положения
3.1. Риск загрязнения материалов и продукции, создаваемый окружающей средой производственных помещений (зданий), должен быть минимальным при условии соблюдения всех мер защиты.
3.2. При эксплуатации помещений следует соблюдать меры предосторожности. При этом проведение технического обслуживания и ремонта не должно оказывать отрицательного влияния на качество продукции. Уборка и дезинфекция помещений должны проводиться в соответствии с письменными инструкциями.
3.3. Освещение, температурный режим, влажность воздуха и вентиляция должны соответствовать назначению помещения и не оказывать прямого или косвенного отрицательного влияния на работу оборудования и лекарственные средства во время их производства и хранения.
3.4. При проектировании и эксплуатации помещений должна быть обеспечена защита от проникания в них насекомых или животных.
3.5. В помещения не должны допускаться лица, не имеющие права доступа в них.
Производственные, складские помещения и помещения контроля качества не должны использоваться для сквозного прохода персонала, не работающего в них.
Производственная зона
3.6. Для минимизации риска для здоровья людей из-за перекрестных загрязнений при производстве некоторых лекарственных средств, таких как сенсибилизирующие вещества (например, пенициллины) или биологические лекарственные средства (например, из живых микроорганизмов), следует предусмотреть специальные и изолированные технические средства (помещения, оборудование, средства обслуживания и др.). В одних и тех же помещениях не допускается производство отдельных видов антибиотиков, некоторых гормонов, цитотоксинов, сильнодействующих лекарственных средств и продукции немедицинского назначения. В исключительных случаях производство таких препаратов допускается в одних помещениях при разделении циклов производства по времени, с соблюдением специальных мер предосторожности и проведением необходимой валидации (аттестации). В зданиях, используемых для производства лекарственных средств, не допускается производство ядов технического назначения (пестицидов и гербицидов).
|
|
|
3.7. Планировочные решения помещений, по возможности, должны соответствовать логической последовательности производственных операций и обеспечивать выполнение требований к чистоте.
3.8. Планировочные решения рабочих зон и зон хранения внутри производства должны обеспечивать последовательное и логичное размещение оборудования и материалов, сводить к минимуму риск перепутывания различных лекарственных средств или их компонентов, перекрестного загрязнения и ошибочного выполнения или пропуска любых операций по производству или контролю.
3.9. Если исходные и первичные упаковочные материалы, промежуточные или нерасфасованные продукты подвергаются воздействию окружающей среды, внутренние поверхности помещений (стены, пол и потолок) должны быть гладкими, не иметь открытых соединений и трещин, не выделять частиц и должны обеспечивать возможность беспрепятственной и эффективной уборки, а также, при необходимости, дезинфекции.
3.10. Конструкция и размещение труб, осветительных приборов, оборудования вентиляции и т.п. не должны иметь мест, труднодоступных для очистки. По возможности их обслуживание должно осуществляться с внешней стороны производственных помещений.
3.11. Трубопроводы для стоков (канализация) должны иметь необходимые размеры и быть оборудованы устройствами, предотвращающими обратный поток. Следует избегать применения открытых желобов. При необходимости они должны быть неглубокими для удобства очистки и дезинфекции.
3.12. В производственных зонах, в зависимости от выпускаемой продукции, выполняемых операций и требований к окружающей среде, следует предусматривать эффективную систему вентиляции с обеспечением требуемой температуры и, при необходимости, влажности воздуха и его фильтрации.
3.13. Исходные материалы взвешивают, как правило, в специально оборудованных для этого помещениях.
3.14. Если выполнение работы сопровождается выделением пыли (например, при отборе проб, взвешивании, смешении, производственных операциях и упаковке сухих продуктов), то необходимо предусмотреть меры по предотвращению перекрестного загрязнения и проведению очистки.
3.15. При проектировании (в т.ч. разработке планировочных решений) помещений для упаковки лекарственных средств следует предусматривать специальные меры, предотвращающие перепутывание или перекрестное загрязнение материалов и продукции.
3.16. Производственные помещения должны быть хорошо освещены, особенно в местах проведения визуального контроля.
3.17. Внутрипроизводственный контроль может проводиться в зоне производства, если это не создает помех для технологического процесса.
Зоны складирования
3.18. Зоны складирования должны иметь достаточную вместимость для обеспечения надлежащего хранения различных категорий материалов и продукции (исходного сырья и упаковочных материалов; промежуточной, нерасфасованной и готовой продукции; продукции, находящейся в карантине; разрешенной для выпуска, отклоненной, возвращенной или отозванной продукции).
3.19. При проектировании и организации зон складирования следует предусматривать надлежащие условия хранения. Зоны складирования должны быть чистыми и сухими, в них должен быть обеспечен требуемый температурный режим. При необходимости следует обеспечивать специальные условия хранения (температура, влажность воздуха и т.п.) и их контроль.
3.20. В зонах приемки и выдачи материалов и продукции должна быть обеспечена их защита от неблагоприятного влияния окружающей среды. Проект зоны приемки должен предусматривать очистку упаковок с поступающими материалами перед их складированием.
3.21. Если режим карантина обеспечивается хранением продукции в раздельных зонах, то эти зоны должны быть четко обозначены. Доступ в них должен быть разрешен только лицам, имеющим на это право. Любая другая система, заменяющая физическое разделение, должна обеспечивать эквивалентную безопасность.
3.22. Отбор проб исходных материалов, как правило, следует выполнять в отдельной зоне. При отборе проб в складской зоне должны быть приняты меры, предотвращающие прямое или перекрестное загрязнение.
3.23. Отклоненные, отозванные или возвращенные материалы и продукцию следует хранить в изолированных зонах.
3.24. Сильнодействующие вещества и препараты должны храниться в безопасных и охраняемых помещениях.
3.25. Должно быть обеспечено надежное и безопасное хранение печатных материалов ввиду их ключевой роли в подтверждении идентичности лекарственных средств.
Зоны контроля качества
3.26. Как правило, лаборатории контроля качества должны быть отделены от производственных помещений. Это особенно важно для лабораторий контроля биологических, микробиологических препаратов или радиоизотопов, которые также должны быть отделены друг от друга.
3.27. Проект контрольных лабораторий должен соответствовать требованиям к выполняемым в них операциям. Площадь лабораторий должна быть достаточной для исключения перепутывания и перекрестного загрязнения, а также для хранения образцов и документации.
3.28. Для размещения чувствительных приборов, нуждающихся в защите от электромагнитных полей, вибрации, повышенной влажности воздуха или других внешних факторов, могут быть предусмотрены отдельные помещения.
3.29. Особые требования предъявляются к лабораториям, в которых проводятся работы с образцами специфических веществ, например, биологическими или радиоактивными материалами.
Вспомогательные зоны
3.30. Комнаты отдыха и приема пищи должны быть отделены от других зон.
3.31. Помещения для переодевания, туалеты и душевые кабины должны иметь удобный доступ; их планировка и размеры должны соответствовать численности персонала. Не допускается выход из туалетов непосредственно в производственные или складские зоны.
3.32. Ремонтные участки должны быть, по возможности, отделены от производственных помещений. При необходимости хранения запасных частей и инструментов в зоне производства должны быть предусмотрены специальные помещения или шкафы.
3.33. Помещения для содержания животных должны быть изолированы от остальных зон, оборудованы отдельными системами вентиляции и отдельным входом.
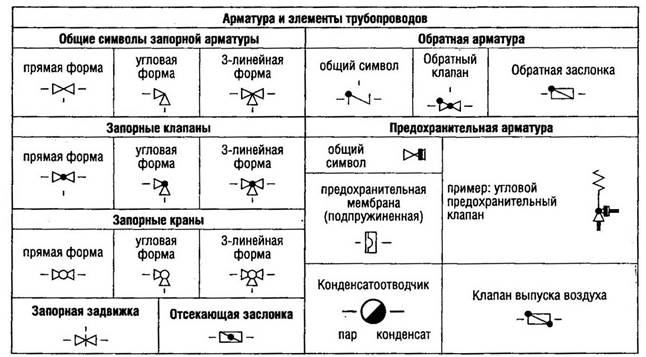
Оборудование
3.34. Конструкция, монтаж и порядок технического обслуживания оборудования должны соответствовать его назначению.
3.35. Работы по ремонту и техническому обслуживанию оборудования не должны оказывать отрицательного влияния на качество продукции.
3.36. Конструкция производственного оборудования должна обеспечивать удобство и возможность его очистки. Операции по очистке оборудования должны выполняться в соответствии с подробными письменными инструкциями. Оборудование следует содержать в сухом и чистом состоянии.
3.37. Инвентарь и материалы для мытья и очистки не должны быть источниками загрязнения.
3.38. Оборудование должно быть установлено так, чтобы, по возможности, исключить риск загрязнения или выполнения ошибочных действий.
3.39. Технологическое оборудование не должно влиять на качество продукции и представлять собой какую-либо опасность для продукции. Части технологического оборудования, контактирующие с продукцией, не должны вступать с ней в химическую реакцию, выделять или абсорбировать вещества, оказывающие влияние на качество продукции.
3.40. Погрешность и диапазон весов и другого измерительного оборудования должна соответствовать производственным и контрольным операциям, в которых они используются.
3.41. Калибровка (поверка) измерительных, регистрирующих, контрольных приборов и весов должна проводиться с установленными интервалами соответствующими методами. Результаты калибровки (поверки) должны быть задокументированы.
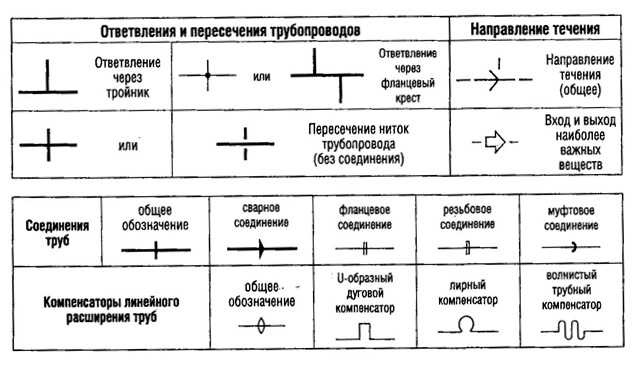
3.42. Стационарные трубопроводы должны иметь маркировку с указанием проходящих по ним веществ и, если требуется, направления потока.
3.43. Трубопроводы для воды очищенной и воды для инъекций (дистиллированной, деионизованной и других видов воды) следует обрабатывать в соответствии с инструкциями, в которых указаны пределы микробного загрязнения и принимаемые меры в случае их превышения.
3.44. Неисправное оборудование следует удалять из зоны производства и контроля качества или обозначать соответствующим образом.
Надлежащая инженерная практика (англ. – Good Engineering Practice (GEP)) определяется как совокупность инженерных методов и стандартов, которые применяются на протяжении жизненного цикла проекта с целью предоставления оптимальных и экономически эффективных решений для соответствия требованиям заказчика и применимым регуляторным требованиям. GEP поддерживает действия по текущему функционированию и будущему планированию фармацевтического бизнеса. Кроме того, документация GEP может быть использована для поддержки работ по верификации производственных систем. Основной целью GEP является предостав ление оптимальных и экономически эффективных инженерных решений для соответствия требованиям заказчика и действующим регулятор ным требованиям.
GEP включает следующие ключевые моменты:
- Профессиональное и компетентное управление проектом (процессы, методы и персонал);
- Слаженный и структурированный проект, закупки у надежных поставщиков, реализация проекта и беспроблемная сдача-приемка;
- Понимание и корректное восприятие:
- потребностей производства в последующей эксплуатации производственных систем;
- законодательных требований в отношении безопасности продукта, здоровья персонала и окружающей среды;
- признанных отраслевых и общесистемных международных стандартов, производственных практик и лучшего опыта;
- Надлежащую документацию по действующему производству и техническому обслуживанию и доказательство соответствия действующим правилам и принципам.
-
- 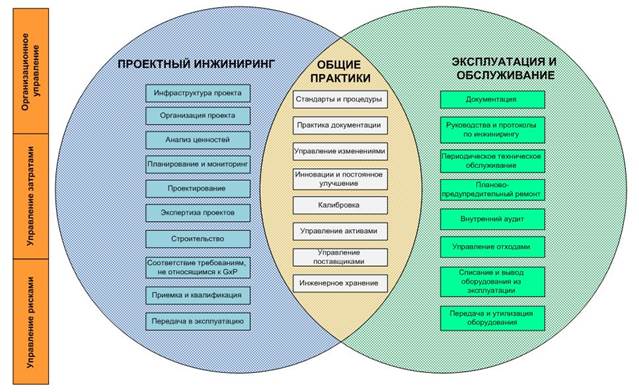
Рис. 1 – Охват и взаимосвязь принципов GEP
|
|
|
