 |


Синдром Эдвардса (трисомия 18)
|
|
|
|
Синдром Патау (трисомия 13)
Синдром Патау выделен в самостоятельную нозологическую форму в 1960 г. в результате цитогенетического обследования детей с врожденными пороками развития. Частота синдрома Патау среди новорожденных равна 1: 5000-7000. Существуют цитогенетические варианты этого синдрома. Простая полная трисомия 13 как следствие нерасхождения хромосом в мейозе у одного из родителей (главным образом у матери) встречается у 80-85% больных. Остальные случаи обусловлены в основном передачей дополнительной хромосомы (точнее, ее длинного плеча) в робертсоновских транслокациях типа D/13 и G/13. Обнаружены и другие цитогенетические варианты (мозаицизм, изохромосома, неробертсоновские транслокации), но они встречаются крайне редко. Клиническая и патолого-анатомическая картина простых трисомных форм и транслокационных форм не различается.
Соотношение полов при синдроме Патау близко к 1: 1. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25-30% ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок гестации 38,3 нед). Характерное осложнение беременности при вынашивании плода с синдромом Патау - многоводие: оно встречается почти в 50% случаев. Синдром Патау сопровождается множественными врожденными пороками развития головного мозга и лица (рис. 5.7). Это патогенетически единая группа ранних (и, следовательно, тяжелых) нарушений формирования головного мозга, глазных яблок, костей мозговой и лицевой частей черепа. Окружность черепа обычно уменьшена, встречается и тригоноцефалия. Лоб скошенный, низкий; глазные щели узкие, переносье запавшее, ушные раковины низко расположенные и дефор-
|
|
|
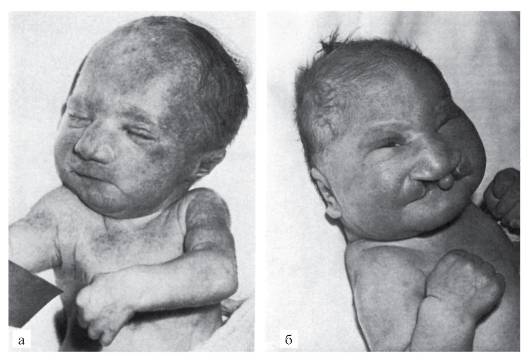
Рис. 5.7. Новорожденные с синдромом Патау (тригоноцефалия (б); двусторонняя расщелина верхней губы и нёба (б); узкие глазные щели (б); низко расположенные (б) и деформированные (а) ушные раковины; микрогения (а); флексорное положение кистей)
мированные. Типичный признак синдрома Патау - расщелины верхней губы и нёба (обычно двусторонние). Всегда обнаруживаются пороки нескольких внутренних органов в разных комбинациях: дефекты перегородок сердца, незавершенный поворот кишечника, кисты почек, аномалии внутренних половых органов, дефекты поджелудочной железы. Как правило, наблюдаются полидактилия (чаще двусторонняя и на руках) и флексорное положение кистей. Частота разных симптомов у детей с синдромом Патау по системам следующая: лицо и мозговая часть черепа - 96,5%, опорно-двигательный аппарат - 92,6%, ЦНС - 83,3%, глазное яблоко - 77,1%, сердечнососудистая система - 79,4%, органы пищеварения - 50,6%, мочевая система - 60,6%, половые органы - 73,2%.
Клиническая диагностика синдрома Патау основывается на сочетании характерных пороков развития. При подозрении на синдром Патау показано УЗИ всех внутренних органов.
В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы жизни (95% умирают до 1 года). Однако некоторые больные живут несколько лет. Более того, в развитых странах отмечается тенденция увеличения продолжительности жизни больных с синдромом Патау до 5 лет (около 15% больных) и даже до 10 лет (2-3% больных).
Другие синдромы врожденных пороков развития (синдромы Меккеля и Мора, тригоноцефалия Опитца) по отдельным признакам совпадают с синдромом Патау. Решающий фактор в диагностике - исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших детей. Точный цитогенетический диагноз необходим для прогноза здоровья будущих детей в семье.
|
|
|
Лечебная помощь детям с синдромом Патау неспецифическая: операции по поводу врожденных пороков развития (по жизненным показаниям), общеукрепляющее лечение, тщательный уход, профилактика простудных и инфекционных болезней. Дети с синдромом Патау практически всегда глубокие идиоты. Продолжительность жизни больных резко снижена. Обычно дети погибают в первые дни или недели жизни
Синдром Эдвардса (трисомия 18)
Почти во всех случаях синдром Эдвардса обусловлен простой трисомной формой (гаметическая мутация у одного из родителей). Встречаются и мозаичные формы (нерасхождение на ранних стадиях дробления). Транслокационные формы крайне редки, и, как правило, это частичные, а не полные трисомии. Клинических различий между цитогенетически различающимися формами трисомии нет..
При синдроме Эдвардса отмечается выраженная задержка пренатального развития при нормальной продолжительности беременности (роды в срок). На рис. 5.8-5.11 показаны пороки при синдроме Эдвардса.Признаки: Это множественные врожденные пороки развития лицевой части черепа, сердца, костной системы, половых органов. Череп долихоцефалической формы; нижняя челюсть и отверстие рта маленькие; глазные щели узкие и короткие; ушные раковины деформированные и низко расположенные. Из других внешних признаков отмечаются флексорное положение кистей, аномальная стопа (пятка выступает, свод провисает), I палец стоп короче II пальца. Спинно-мозговая

Рис. 5.8. Новорожденный с синдромом Эдвардса (выступающий затылок, микрогения, флексорное положение кисти)

Рис. 5.9. Характерное для синдрома Эдвардса положение пальцев (возраст ребенка 2 мес)

Рис. 5.10. Стопа-качалка (пятка выступает, свод провисает)

Рис. 5.11. Гипогенитализм у мальчика (крипторхизм, гипоспадия)
грыжа и расщелина губы встречаются редко (5% случаев синдрома Эдвардса).
Многообразная симптоматика синдрома Эдвардса у каждого больного проявляется лишь частично: лицо и мозговая часть черепа - 100%, опорно-двигательный аппарат - 98,1%, ЦНС - 20,4%, глаза - 13,61%, сердечно-сосудистая система - 90,8%, органы пищеварения - 54,9%, мочевая система - 56,9%, половые органы - 43,5%.
Как видно из представленных данных, наиболее значимы в диагностике синдрома Эдвардса изменения мозгового черепа и лица, опорно-двигательного аппарата, пороки развития сердечнососудистой системы.
|
|
|
Дети с синдромом Эдвардса умирают в раннем возрасте (90% до 1 года) от осложнений, обусловленных врожденными пороками развития (асфиксии, пневмонии, кишечной непроходимости, сердечно-сосудистой недостаточности). Клиническая и даже патолого-анатомическая дифференциальная диагностика синдрома Эдвардса сложна, поэтому во всех случаях показано цитогенетическое исследование. Показания для него те же, что и при трисомии 13 (см. выше).
|
|
|
