 |


Электронное строение атомов углерода. Виды гибридизации.
|
|
|
|
Основы строения органических соединений
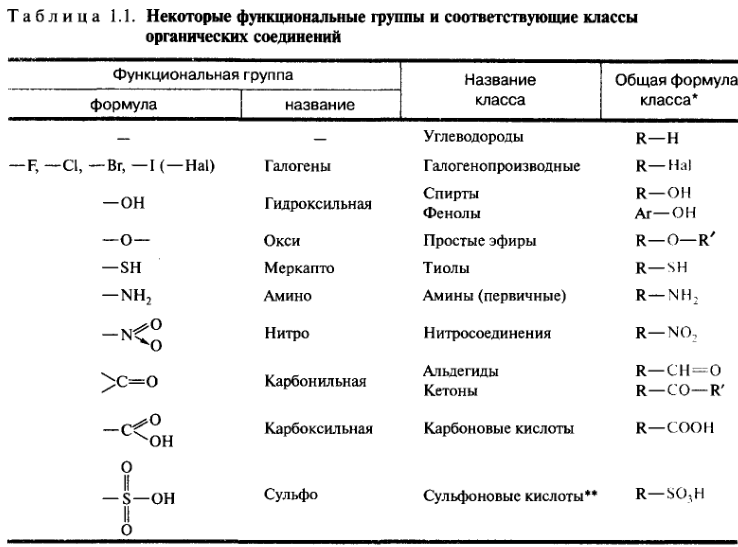
Классификация органических соединений. Функциональная группа и строение углеродного скелета как классификационные признаки органических соединений, Главные классы органических соединений.
В основу современной классификации органических соединений положены два важнейших признака:
• строение углеродного скелета молекулы;
• наличие в молекуле функциональных групп.
По строению углеродного скелета органические соединения делятся на группы. Ациклические (алифатические) соединения, в которых цепь атомов углерода может быть неразветвленной или разветвленной. Карбоциклические соединения, в которых цепь, состоящая только из атомов углерода, замкнута в цикл (кольцо). Гетероциклические соединения, имеющие в составе циклического скелета, кроме атомов углерода, один или несколько гетероатомов — как правило, атомы азота, кислорода или серы:

Родоначальными соединениями в органической химии считаются углеводороды, состоящие только из атомов углерода и водорода. В большинстве своем органические молекулы содержат функциональные группы, т. е. атомы или группы атомов, определяющие химические свойства соединения и принадлежность его к определенному классу. В состав функциональной группы обязательно входит гетероатом, хотя иногда к функциональным группам причисляют и углерод-углеродные кратные связи (С=С и С≡С). Многие такие группы вообще не содержат атом углерода. В зависимости от наличия в молекуле тех или иных функциональных групп органические соединения делятся на классы.
| Функциональная группа | Название класса | Общая формула класса |
| -R | углеводороды | R-H |
| -Hal | галогенопроизводные | R-Hal |
| -OH | спирты, фенолы | R-OH Ar-OH |
| -О- | простые эфиры | R-O-R` |
| -SH | тиолы | R-SH |
| -NH2 | амины | R-NH2 |
| -NO2 | нитросоединения | R-NO2 |
| >C=O | альдегиды кетоны | R-CHO R-CO-R |
| -COOH | карбоновые кислоты | R-COOH |
| -SO3H | сульфокислоты | R-SO3H |
Соединения, имеющие в молекуле одну функциональную группу, называются монофункциональными; несколько одинаковых функциональных групп – полифункциональными (глицерин). Гетерофункционалъные соединения содержат в молекулах различные функциональные группы. Их можно одновременно отнести к нескольким классам.
|
|
|
Переход от одного класса к другому осуществляется чаще всего с участием функциональных групп без изменения углеродного скелета. Кроме того, классификационные признаки положены в основу номенклатуры органических соединений.
Номенклатура органических соединений. Тривиальная номенклатура. Основные принципы номенклатуры IUPAC (IUPAC-Международный союз теоретической и прикладной химии): заместительная и радикало -функциональная номенклатуры.
Номенклатура должна быть систематической и международной, чтобы могли отобразить в названии структуру соединения и по названию однозначно представить структуру. Кроме того, номенклатура должна быть пригодной для компьютерной обработки.
Исторически первыми были тривиальные названия веществ, которые указывали либо на источник выделения (кофеин, мочевина), либо свойства веществ (глицерин, глюкоза). Широко распространены торговые названия, причем для лекарственных веществ часто в основу такого названия берется фармакологический эффект или отдельные элементы структуры. Эти названия удобны своей лаконичностью, но они не дают представления о строении вещества и не могут быть объединены в систему. К тому же некоторые из тривиальных названий со временем выходят из употребления, хотя многие из них прочно вошли в обиход и даже легли в основу систематических названий.
|
|
|
Использование систематической номенклатуры применительно к лекарственным веществам играет важную роль в фармации, поскольку многие лекарства выпускаются под разнообразными торговыми названиями. При переводе же их в систематические можно зачастую убедиться, что действующим началом этих лекарственных средств может оказаться одно и то же вещество (парацетамола, панадола, тайленола – n-гидроксиацетанилид). В ходе развития органической химии возникали различные номенклатурные системы (Женевская, 1892; Льежская, 1930), которые после многократных усовершенствований стали основой современной систематической номенклатуры ИЮПАК (IUPAC — Международный союз теоретической и прикладной химии).
Номенклатура органических соединений — это система терминов, обозначающих строение веществ и пространственное расположение атомов в их молекулах.
Систематическое название — полностью составленное из специально созданных или выбранных слогов, (пентан, тиазол). Тривиальное название — в котором ни один из слогов не используется в систематическом смысле (мочевина, фуран). Родоначальное название — та часть названия, от которой по определенным правилам строится название целиком. Например, «этан» – «этанол». Может быть как систематическим, так и тривиальным.
Заместитель — любой атом или группа атомов, замещающие в исходном соединении атом водорода.
Характеристическая группа — в ИЮПАК практически эквивалентен понятию «функциональная группа», например: аминогруппа, галогены, гидроксильная группа, карбоксильная группа, карбонильная группа, оксогруппа, нитрогруппа, цианогруппа. Старшая (главная) группа — характеристическая группа, название которой отражается суффиксом. Никаких других преимуществ не имеет.
Умножающие префиксы — приставки ди-, три-, тетра- и т. д., применяемые для обозначения числа одинаковых заместителей или кратных связей. Локант — цифра или буква, указывающая положение заместителя или кратной связи в родоначальном названии.
Из восьми типов номенклатур в ИЮПАК наиболее универсальной и распространенной является заместительная номенклатура. Реже используется радикально-функциональная номенклатура.
|
|
|
Заместительная номенклатура. Название строится как сложное слово, состоящее из корня (родоначальное название), префиксов и суффикса, характеризующих число и характер заместителей, степень ненасыщенности; указываются локанты. Характеристические группы делятся на два типа. Одни из них обозначаются только в виде префиксов, другие могут быть суффиксами или префиксами в зависимости от старшинства. За старшую принимают ту группу, которая находится выше других в табл. Все другие обозначаются префиксами.



Радикально-функциональная номенклатура. Для названий в основном используются те же принципы, но для отражения старшей группы никогда не применяются суффиксы. Вместо этого одним словом отражают название функционального класса, а остальную часть названия — соответствующим радикалом. Для двухвалентных характеристических групп указывают оба радикала, связанные с этой группой. Если соединение включает более одного типа характеристических групп, то за название функционального класса принимают такое, которое расположено выше других в табл. Остальные группы префиксами.

Принципы построения систематических названий. Включает следующие:
1. Определяют тип номенклатуры, который целесообразно применить к данному конкретному соединению.
2. Определяют старшую характеристическую группу. Именно она обусловливает в дальнейшем выбор родоначальной структуры и ее нумерацию.
3. Определяют родоначальную структуру — главную углеродную цепь или основную циклическую систему, которая должна включать максимальное число старших групп. Главная углеродная цепь для ациклических соединений выбирается по критериям, при этом каждый последующий критерий вступает в действие лишь тогда, когда предыдущий не приводит к выбору:
а) максимальное число старших групп;
б) максимальное число кратных (двойных и тройных) связей;
в) максимальная длина цепи;
г) максимальное число заместителей.
4. Называют родоначальную структуру и старшую характеристическую группу.
|
|
|
5. Определяют и называют заместители.
6. Проводят нумерацию так, чтобы старшая группа получила наименьший номер. Если выбор неоднозначен, то применяют правило наименьших локантов — нумеруют так, чтобы заместители получили наименьшие номера. Наименьшая последовательность та, в которой первой встретится цифра меньшая, чем в другой последовательности (1,2,7- < 1,3,4 -).
7. Объединяют отдельные части названия в общее, придерживаясь алфавитного порядка префиксов (умножающие приставки не включаются). Цифры-локанты ставят перед префиксами и после суффиксов.
8. Радикально-функциональная номенклатура лишь там, где она традиционно используется до настоящего времени.
Допускаются несистематические названия для следующих незамещенных углеводородов изостроения: изобутан (СН3)2СНСН3, изопентан (СН3)2СНСН2СН3, неопентан (СН3)4С, изогексан (СН3)2СНСН2СН2СН3. Для ненасыщенных соединений: этилен СН2=СН2, ацетилен СН≡СН, аллен СН2=С=СН2, изопрен СН2=С(СН3)СН=СН2.
В ряду ароматических углеводородов — аренов, сохраняются следующие несистематические названия:

Из родоначальных структур конденсированных аренов наиболее часто встречаются четыре. В ряде случаев сохраняется исторически сложившаяся нумерация (например, антрацен и фенантрен).

Несложные по структуре галогенопроизводные часто называют по радикально-функциональной номенклатуре, например, изопропилбромид (СН3)2СНВг, бензилхлорид С6Н5СН2С1.
Тривиальные названия сохраняются для ряда многоатомных спиртов и фенолов:
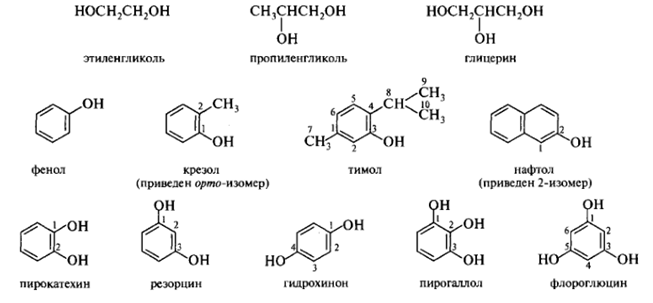
Менее употребительны радикально-функциональные названия солей спиртов, образуемые заменой части названия -иловый спирт на суффикс -илат, например, этилат натрия C2H5ONa, триизопропилат алюминия [(СН3)2СНО]3А1.
Для простых эфиров, чаще чем для других классов соединений, применяется радикально-функциональная номенклатура. В этом случае названия образуют из названий радикалов R и R' в алфавитном порядке, предшествующих слову эфир, например, метилэтиловый эфир СН3—О—СН2СН3, диизо- пропиловый эфир (СН3)2СН—О—СН(СН3)2, винилфениловый эфир С6Н5—О—СН=СН2.
Некоторые амины сохраняют тривиальные названия:

Если соответствующая альдегиду карбоновая кислота имеет тривиальное название (1.3.10), то из него может быть образовано и тривиальное название альдегида:

Сохраняются следующие тривиальные названия:

Сохраняется тривиальное название «ацетон» для СН3СОСН3. Для многих алифатических и карбоциклических карбоновых кислот сохранены тривиальные названия, обычно предпочтительнее систематических.
|
|
|
Электронное строение атомов углерода. Виды гибридизации.
Строгое рассмотрение понятия химической связи базируется на принципах квантовой механики. Фундаментальный принцип квантовой механики гласит, что электроны ведут себя как волны и движение электрона можно описать с помощью волновой функции. Математическая модель электронов в атоме известна как уравнение Шрёдингера. Решение дифференциального уравнения Шрёдингера позволяет получить характеристику энергетических уровней и соответствующие волновые функции, описывающие движение электронов в атоме. Квадрат модуля волновой функции всегда положительный. Он соответствует плотности электронного облака в данном объеме. Графические трехмерные изображения электронной плотности называются орбиталями.
Атомной орбиталью (АО) называется область пространства, в котором вероятность нахождения электрона максимальна.
Состояние электрона в атоме оценивается с помощью квантовых чисел, которые характеризуют энергетический уровень, форму и пространственную направленность орбитали. Для объяснения строения электронных оболочек атомов привлекаются три основных положения: принцип Паули, правило Гунда и принцип минимума энергии. Атомы и молекулы являются типичными примерами квантово-механических систем. При сближении атомов происходит перекрывание их АО. Молекула описывается распределением электронов между наборами молекулярных орбиталей (МО). Существуют три безразмерных квантовых числа, которые обозначают символами n, l и m. Появление квантового числа n вызвано тем, что электрон может менять свое расстояние от ядра. Квантовые числа l и m связаны с угловым моментом количества движения электрона, который может вращаться вокруг ядра в трех измерениях. Число l характеризует величину углового момента, а число m - ориентацию углового момента в пространстве, так как угловой момент - векторная величина. Число n называется главным квантовым числом. Допустимыми значениями квантовых чисел, которые вытекают из граничных условий, являются n = 1, 2, 3...; l = 0, 1, 2... (n-1); m = l, (l-1), (l-2),..., -l.
Все орбитали с нулевым угловым моментом называются s-орбиталями. s-Орбиталь низшей энергии (n=1, l=0, m=0) называется 1s-орбиталью. Если n=2 и l=0, то это 2s-орбиталь. Если n=0, единственным значением, разрешенным для l, является нуль, но если n=2, квантовое число орбитального углового момента может принимать значения 0 (2s-орбиталь) или 1. Если l=1, атомные орбитали носят название р-орбиталей. При n=2 и l=1 мы имеем 2р-орбиталь. Поскольку для р-орбиталей l=0, квантовое число m может принимать значения +1, 0 и -1. Разные значения m соответствуют орбиталям с различными ориентациями орбитального углового момента. р-Орбиталь с m=0 имеет нулевую проекцию углового момента на ось z, и по этой причине ее называют pz-орбиталью. Две другие р-орбитали можно представить аналогичными картинами с ориентацией «лопастей» вдоль осей x и y, поэтому они называются px- и py-орбиталями. Если n=3, то l может принимать значения 0, 1 и 2. Это приводит к одной 3s-орбитали, трем 3р-орбиталям и пяти 3d-орбиталям. 3d-Орбиталей пять, поскольку при l =2 m может принимать значения 2, 1, 0, -1 и -2.
Чтобы отличить друг от друга два электрона на s-орбитали, необходимо еще одно квантовое число, которое называется спином. Спин связан с угловым моментом электрона, вращающегося вокруг собственной оси. Для электрона возможно лишь одно значение s=1/2. Единственное различие между двумя электронами на s-орбитали заключается в различной ориентации спинового углового момента. Таким образом, из двух электронов на 1s-орбитали один имеет α-спин, а другой - β-спин, т.е. спины этих электронов антипараллельны или, по-другому, спарены.
Существует еще один важный принцип квантовой теории, который запрещает занимать какую-либо орбиталь более чем двум электронам. Этот принцип называется запретом Паули: любая орбиталь может быть занята не более чем двумя электронами, и если ее занимают два электрона, направление их спинов должно быть противоположным. Запрет Паули относится как к атомным, так и к молекулярным орбиталям. Принцип Паули запрещает, чтобы третий электрон находился на уже заполненной двумя электронами s-орбитали, и поэтому третий электрон занимает следующую орбиталь низшей энергии.
Чтобы построить электронную конфигурацию любого атома с номером Z, нужно представить себе атомные орбитали с последовательностью энергий 1s<2s<2p<3s<3p<3d<... и затем разместить Z электронов, начиная с орбитали низшей энергии, в соответствии с принципом Паули. Необходимо лишь помнить, что имеется только одна 1s-орбиталь, одна 2s-орбиталь и т.д., но орбиталей типа 2р, 3р и т.д. по три, орбиталей типа 3d, 4d и т.д. - по пять, а орбиталей типа 4f, 5f и т.д. - по семь.
Согласно понятию гибридизации, четыре валентные орбитали атома углерода 2s, 2рх, 2pz,2рz, могут быть заменены набором из определенного числа эквивалентных гибридных орбиталей. Следует помнить, что гибридизация — это не физическое явление, а чисто математический прием. В зависимости от комбинации гибридных и негибридизованных орбиталей атом углерода может находиться в состоянии sp3-, sp2- или sp-гибридизации. Представление об sp3-гибридизации атома углерода можно описать следующим образом.

Для перехода электрона с 2s- на 2р-орбиталь требуется небольшое количество энергии, которое легко компенсируется энергией, высвобождаемой при образовании двух дополнительных связей.
Оперируя понятием гибридизации, можно объяснить равноценность всех четырех химических связей в метане. Кроме того, гибридные орбитали способны к лучшему перекрыванию. Если принять относительную эффективность перекрывания s-AO за единицу, то, согласно расчетным данным, эффективность перекрывания других орбиталей возрастает в последовательности:
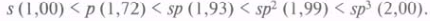
Др. типы гибридизации далее.
Таким образом, концепция гибридизации позволяет определить, где в пространстве локализованы молекулярные орбитали, т.е. связывает классические и квантовомеханические представления о структуре соединений.
4. Типы химических связей в органических соединениях. Ковалентные s– и p–связи. Строение двойных (С=С, С=О, С=N) и тройных (СºС, CºN) связей, их основные характеристики (длина, энергия, полярность, поляризуемость).
Молекулы органических соединений представляют собой совокупность атомов, связанных в определенной последовательности химическими связями. Реакционная способность соединений обусловлена типом химических связей, природой связываемых атомов и их взаимным влиянием в молекуле.
Химическая связь — совокупность взаимодействий между электронами и ядрами, приводящих к соединению атомов в молекулу.
Локализованная связь — это химическая связь, электроны которой поделены между ядрами двух атомов. Для органических соединений характерны ковалентные s - и p -связи. Ковалентная связь — это химическая связь, образованная за счет обобществления электронов связываемых атомов.
Ковалентная связь образуется в результате перекрывания двух АО с образованием молекулярной орбитали, занимаемой двумя электронами. Л. Полинг ввел полезные для понимания ковалентной связи понятия направленной валентности и гибридизации орбиталей. Согласно понятию направленной валентности, связь атомов осуществляется в том направлении, при котором обеспечивается максимальное перекрывание орбиталей. Чем лучше перекрывание, тем прочнее должна быть связь, и только при максимальном перекрывании достигается минимум энергии системы.

При образовании ковалентных связей посредством перекрывания р-орбиталей доли р-орбиталей помечают «+» и «-», (не соотносить с зарядами). Обе доли р -электронного облака несут отрицательный заряд, но волновая функция всегда имеет противоположные знаки по обе стороны узла орбитали. Перекрываются орбитальные доли одинакового знака. Типы перекрывания орбиталей могут характеризоваться цилиндрической симметрией относительно межъядерной оси, что отвечает понятию s -связи.
s-Связь — это одинарная ковалентная связь, образованная при перекрывании АО по прямой (оси), соединяющей ядра 2х связываемых атомов с максимальным перекрыванием на этой прямой.

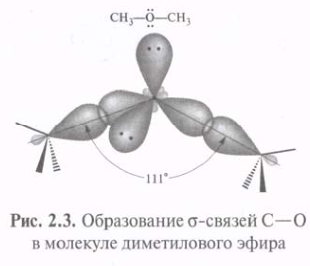
Использование sp3-гибридных орбиталей в связывании атома 12С с четырьмя атомами 1Н при образовании молекулы СН4 приводит к возникновению более прочных s -связей С—Н. Метан с четырьмя идентичными заместителями у атома углерода представляет собой идеальный тетраэдр с углом Н—С—Н, равным 109°28'. Такая геометрия обеспечивает минимальное отталкивание между 4-мя связывающими парами электронов. Атомы 16О, 14N и др., подобно 12С, могут использовать sp3-гибридные орбитали для образования прочных s -связей.
В этилене каждый из атомов углерода связан не с 4мя, а только с 3мя другими. В данном случае электронное строение молекулы описывается с привлечением представлений об sp2-гибридизации. Три sp2-АО, образовавшиеся из одной 2s- и двух 2р-орбиталей, лежат в одной плоскости под углом 120°. В этилене s -связь С—С образуется путем перекрывания гибридных орбиталей вдоль их осей. Две оставшиеся sp2-орбитали каждого из атомов углерода перекрываются с s-AO водорода, образуя s -связи С—Н. Экспериментально установлено, что углы между связями Н—С—Н и Н—С—С составляют соответственно 116,7° и 121,6°, т. е. наблюдается некоторое отклонение от идеального угла 120°. Негибридизованная 2р-АО располагается под прямым углом к плоскости каркаса σ-связей. Параллельные друг другу 2р-АО двух атомов углерода перекрываются над и под плоскостью σ-скелета с образованием МО π-связи (рис. 2.4, в).
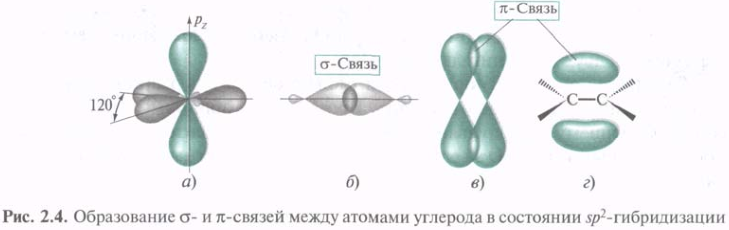

π-Связь — это связь, образованная при боковом перекрывании негибридизованных р-АО с максимальным перекрыванием над и под плоскостью σ-связей. Электронная плотность π-связи концентрируется выше и ниже плоскости σ-связей. Плоскость, проходящая через ядра, представляет собой узловую плоскость. Вероятность нахождения в этой плоскости π-электронов равна нулю.
Представление об sp2-гибридизации можно применить и для 16О, 14N, Hal. При образовании двойной C=N 14N использует 1 гибридную орбиталь для перекрывания с sp2-АО 12С с образованием σ-связи, другую — для σ-связи др. атомом, а 3-я занята неподеленной парой электронов. При этом и у 12С и у 14N остаются негибридизованные р-орбитали, которые посредством бокового перекрывания образуют π-связь. Аналогично образуется связь С=O с той разницей, что на 2-х гибридных орбиталях 16О располагаются две пары электронов.
Для описания связи С=О в карбонильной группе можно применить и представление об sp-гибридизации 16О. В этом случае две неподеленные пары электронов 16О располагаются на неэквивалентных орбиталях: одна на sp-гибридной АО, другая — на рy-АО, перпендикулярной p-орбиталям π-связи С=O.
В алкинах каждый 12С тройной связи С≡С может быть связан только с 2-мя др.. В ацетилене оба 12С находятся в состоянии sp-гибридизации. Гибридные орбитали расположены на одной прямой под углом 180°. При образовании С≡С гибридные орбитали 12С участвуют в построении σ-связи. Две негибридизованные р-орбитали каждого из двух 12С параллельны друг другу и могут попарно перекрываться. При этом образуются две π-связи в перпендикулярных плоскостях. Представление об sp-гибридизации используется и при описании тройной связи 12С с 14N. Неподеленная пара электронов 14N располагается на sp-АО.

Атомы 14N, 16О, серы и фосфора при образовании обычных ковалентных связей используют не все внешние валентные электроны. На гибридных или негибридизованных орбиталях у них имеются одна или более неподеленных пар электронов. При взаимодействии заполненной двухэлектронной АО такого гетероатома (донора) с вакантной орбиталью атома, имеющего недостаток электронов (акцептора), образуется новая ковалентная связь.
Донорно-акцепторная, или координационная, связь — это ковалентная связь, образованная за счет пары электронов одного атома. Например, донорно-акцепторная связь образуется в результате взаимодействия аминов с протонами кислот, при этом два электрона донора в одинаковой степени принадлежат двум связанным атомам. Атом-донор в результате приобретает положительный заряд. Образовавшаяся ковалентная связь, например, в ионе алкиламмония, отличается от других только способом образования, по свойствам идентична другим N—Н.
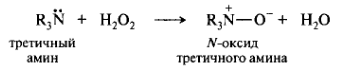
Разновидностью донорно-акцепторной связи является семиполярная связь. Семиполярная связь является сочетанием ковалентной и ионной связей. В этом случае атом-донор образует связь с нейтральным атомом, у которого для завершения внешней валентной оболочки недостает пары электронов. Например, такая связь образуется в N-окcидах при взаимодействии аминов с H2O2. 14N предоставляет пару электронов для образования связи с атомом 16О. В результате ковалентного связывания происходит перераспределение электронной плотности, и на связанных атомах возникают противоположные по знаку заряды. Характерным признаком семиполярной связи служит наличие противоположных зарядов на ковалентно-связанных атомах.
К донорно-акцепторному типу связей относятся также связи в комплексных соединениях. Донором пары электронов могут быть гетероатом с неподеленной парой электронов (n-доноры) или π-электроны изолированной π-связи или системы π-связей (π-доноры). Акцепторами могут служить ионы Me (имеющие вакантные орбитали), молекулярные иод, бром (за счет расширения внешней валентной оболочки), электронодефицитные π-системы (соединения, в которых π-связь или π-система обеднена электронной плотностью из-за акцепторного влияния заместителей). Например, диоксан-триоксид серы.
Особым случаем являются металлоцены — π-комплексы ароматического циклопентадиенид-иона с ионами переходных металлов (Fe2+, Со2+, Ni2+). В ферроцене взаимодействие двух колец циклопентадиенидионов с ионом Fe2осуществляется за счет перекрывания связывающих π-МО богатых электронной плотностью колец с вакантными 3d-АО иона Fe2+.

Свойства ковалентной связи выражаются через ее количественные характеристики — длину, энергию, полярность, поляризуемость.
Длина связи — это расстояние между центрами связанных атомов. Основными методами определения длин связей и углов между ними служат рентгеноструктурный анализ (для твердых) и электронография (для газообразных). Атомы в молекуле колеблются относительно некоторого оптимального расстояния — равновесной длины связи, соответствующей минимуму энергии системы из двух ядер. Поэтому расстояния — это средние значения. Длины связей зависят от природы связи, но одинаковые по типу между одними и теми же атомами связи в разных соединениях имеют приблизительно постоянное значение (свойства отдельных связей в приближении не зависят от остальной части молекулы).
Длины связей с участием атома углерода зависят от его состояния гибридизации. Одинарные связи С—С имеют тенденцию к уменьшению длины с увеличением доли s-характера гибридной орбитали. Так, длины связей Csp3—Csp2, Csp2—Csp2, Csp3—Csp равны 0,154, 0,150 и 0,146 нм. Ту же тенденцию можно отметить и для связей С—Н: Csp3—Н > Csp2—Н > Csp— Н (0,110; 0,107 и 0,106 нм). При увеличении кратности связей между атомами их длина всегда уменьшается. Двойные связи С=С, С=O, C=N короче соответствующих одинарных, а тройные связи С≡С, C≡N короче соответствующих двойных.
Половина длины ковалентной связи между одинаковыми атомами в молекуле называется ковалентным радиусом. В случае, когда ковалентно связаны разные атомы и радиус одного атома известен, то, определив длину связи, можно вычислить ковалентный радиус другого атома: длина ковалентной связи равна сумме ковалентных радиусов связанных атомов. Исключение составляют сильно полярные связи: их длина меньше, чем сумма ковалентных радиусов.
Еще одной характеристикой расстояний между атомами служит ван-дер-ваальсов радиус, являющийся мерой того, насколько могут сблизиться друг с другом два атома, не связанные ковалентно. Он всегда больше, чем ковалентный.
Валентные углы — это углы между двумя связями, имеющими общий атом. Углы межъядерных связей X—С—Y в органических соединениях должны соответствовать состоянию гибридизации атома углерода и быть равными 109,5, 120 и 180° для sp3-, sp2-, sp-гибридного состояния соответственно. Когда атом углерода в состоянии sp3-гибридизации связан с 4-мя одинаковыми атомами или группами, валентные углы соответствуют углам правильного тетраэдра. Но в большинстве они отличаются от идеальных. Для атомов углерода в состоянии sp2- и sp -гибридизации, связанных с неодинаковыми заместителями, также наблюдаются отклонения от углов 120 и 180° соответственно. Особенно это касается атомов или групп атомов, имеющих разную электроотрицательность. Пространственные затруднения также влияют на изменение валентных углов.
Энергия связи — это та энергия, которую необходимо затратить для разрыва связи между двумя атомами, и соответственно эта же энергия выделяется при образовании связи. Энергию связи можно определить с помощью спектральных и термохимических методов. Энергия служит мерой прочности связи: чем больше энергия, тем связь прочнее.
Энергия, необходимая для гомолитического расщепления связи на атомы, называется энергией диссоциации. Для двухатомных молекул она равна энергии связи. Энергию диссоциации можно измерить, но в случае сложных молекул часто бывает невозможно определить энергию диссоциации, необходимую для разрыва отдельной связи. Обычно энергию, необходимую для превращения молекул в атомы, рассчитывают по теплоте сгорания, исходя из предположения аддитивности вкладов каждого элемента.
Имеется корреляция между длиной связи и ее энергией: чем длиннее связь, тем меньше энергия и наоборот. Двойные связи прочнее и короче, чем соответствующие одинарные связи, но прочность их не вдвое больше. Это означает, что σ-связь прочнее π-связи. Энергия связи может заметно изменяться в зависимости от ряда факторов, связанных со структурными особенностями. Так, энергия связи С—Н для первичного, вторичного и третичного атома углерода не одинакова. Связь с участием третичного атома углерода наименее прочная, с участием первичного — наиболее прочная.
Полярность связи обусловлена неравномерным распределением электронной плотности. Если атомы, образующие ковалентную связь, равноценны, то пара электронов связи в равной степени принадлежит обоим. Большинство же ковалентных связей образовано неодинаковыми или неравноценными атомами. В этом случае электронная плотность может быть смещена. Склонность атомов притягивать электроны характеризуется эмпирическим критерием — электроотрицательностью — это способность атома в молекуле притягивать валентные электроны, участвующие в химической связи.
Были предприняты попытки дать количественную оценку электроотрицательности, которая указывала бы направление и степень смещения электронного облака между любыми двумя атомами. Наиболее известна шкала Л. Полинга (1939) на основе энергий связи двухатомных молекул. В некоторых подходах рассчитывалась электроотрицательность для различных состояний гибридизации атома. Известно, что увеличение доли s-орбитали в гибридной АО приводит к увеличению электроотрицательности. Кроме того, рассчитаны электроотрицательности не только для атомов, но и для групп атомов.
Связь, образованная разными по электроотрицательности атомами, будет полярной. Атомы, связанные ей, несут частичные заряды, обозначаемые δ (дельта). При разности в электроотрицательности атомов связи от 0,5 до 2,0 говорят о сильнополярной связи; если эта разность больше 2,0, то велика степень ионности связи. Смещение электронной плотности полярной σ-связи обозначают прямой стрелкой, совпадающей с валентной чертой, смещение полярной кратной связи - изогнутой стрелкой.
Неравномерное распределение электронной плотности ковалентной связи создает разделение зарядов, характеризуемое дипольным моментом μ. Суммарный дипольный момент молекулы определяют экспериментально. Дипольный момент отдельной связи можно непосредственно измерить только для двухатомных молекул. Молекула более сложного состава рассматривается как система нескольких диполей. Суммарный дипольный момент молекулы является векторной суммой моментов связей. В симметрично построенных молекулах (СС14 или СО2) μ = 0, хотя связи характеризуются значительным дипольным моментом. Однако они компенсируют друг друга. Полярность связей в значительной степени определяет реакционную способность и механизм реакций органических соединений.

Поляризуемость связи выражается в смещении электронного облака по отношению к ядрам под влиянием внешнего электромагнитного поля. Возникающий при этом индуцированный диполь складывается с постоянным диполем (если он есть). Поляризуемость определяется легкостью смещения электронов связи. Легче поляризуются те связи, максимум электронной плотности которых располагается дальше от связываемых ядер. По поляризуемости π-связь значительно превосходит σ-связь. Поляризуемость в значительной мере определяет реакционную способность молекул, так как смещение электронов тех или иных связей может происходить не только под влиянием электрического поля, но и под влиянием приближающейся реагирующей частицы, а также под влиянием растворителей.
Атом водорода, связанный с сильно электроотрицательным атомом (фтора, кислорода, азота, хлора), способен взаимодействовать с неподеленной парой электронов другого сильно электроотрицательного атома этой же или другой молекулы с образованием дополнительной слабой связи, называемой водородной связью.
Электронное облако связи 1Н с электроотрицательным атомом сильно смещается в сторону этого атома, оставляя ядро 1Н слабо экранированным. Большой положительный заряд ядра атома 1Н притягивается отрицательным зарядом другого электроотрицательного атома. Энергия такого взаимодействия соизмерима с энергией прежней связи, и 1Н связывается сразу с двумя, причем связывание со вторым атомом может быть даже более прочным. В результате протон может переходить от одного электроотрицательного атома к другому. Энергетический барьер такого перехода невелик. Природа водородной связи имеет электростатический и донорно-акцепторный характер. Водородная связь слабая, лежит в пределах 10—40 кДж/моль, что значительно меньше энергии ковалентной или ионной связи.
Водородная связь играет значительную роль в проявлении многих физических и химических свойств молекул. Межмолекулярные водородные связи обусловливают ассоциацию многих соединений, например, спиртов, карбоновых кислот, что приводит к аномально высоким температурам их кипения. Сольватация веществ посредством образования водородных связей с растворителем резко повышает их растворимость. Водородные связи также вносят вклад в стабилизацию ионизированных частиц в растворе. Внутримолекулярные водородные связи образуются в том случае, когда возможно замыкание шестичленного и реже пятичленного цикла. Водородные связи играют важнейшую роль в формиро
|
|
|
