 |


Антиоксидантные механизмы клеток
|
|
|
|
Антиоксиданты — это молекулы, обладающие лабильным водородным атомом с неспаренным электроном:
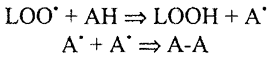
где АН — антиоксидант, А-А его стабильный несвободнорадикальный продукт.
Множество антиоксидантов, вырабатываемых клетками и поглощаемых извне в качестве полностью, либо частично незаменимых соединений сдерживают клеточное «атомное оружие», препятствуя длительному существованию высоких концентраций АКР. Кроме того, избыток АКР может секвестрироваться в пероксисомах. Антиоксиданты — не просто набор веществ. Они способны восстанавливать друг друга и представляют собой антиоксидантные системы клеток.
Основными разрушителями АКР служат ферменты каталаза, супероксиддисмутаза, глютатионпероксидаза, фосфолипид-глютатионпероксидаза и глютатионредуктаза.
По П.Хорнсби и Дж.Кривелло, имеются три класса антиоксидантов (1983):
Каталаза и глютатионпероксидаза — это энзимы предупредительного действия поскольку они восстанавливают АКР (перекись водорода), провоцирующую цепной свободно-радикальный процесс, до неактивного состояния, Супероксиддисмутаза — фермент-прерыватель цепной реакции. Она превращает при [190] наличии восстановительных эквивалентов, супероксидный анион, способный, как показано выше, формировать наиболее активные АКР, в менее активную перекись водорода, разрушаемую каталазой. Субстратами — прерывателями цепной реакции служат фенолы (например, токоферол) и амины (например, цистамин).
Третья разновидность антиоксидантов — хелатирующие агенты, способные связывать железо и другие металлы-катализаторы и разветвители цепных свободнорадикальных реакций (например, десферол и унитиол).
Все упомянутые энзимы и их изоэнзимы являются металлоферментами. В состав их активных центров входят микроэлементы.
|
|
|
Глутатионпероксидаза и фосфолипид-глутатионпероксидаза, утилизующие этот метаболит для инактивации перекиси водорода и липоперекисей с образованием спиртовой группы — это селеносодержащие ферменты. Различные тканевые изоферменты супероксиддисмутазы содержат цинк, или марганец и медь. Митохондриальная изоформа использует марганец, а цитозольные — цинк и медь.
Каталаза является пероксисомальным железозависимым металлоферментом.
Главные антиоксидантные субстраты клеток — это тиоловые соединения.
К ним относятся глютатион, цистеин, Д-пеницилламин.
Глютатион — важнейший компонент антиоксидантных систем печени, сердца, мозга, легких и клеток крови. Показана его защитная роль для эритроцитов — при гемолизе, для нейронов — при инсульте и нейродегенеративных заболеваниях, для альвеоцитов — при отравлении озоном, для кардиомиоцитов — при миокардиодистрофиях. Глютатион обладает радиопротекторными свойствами.
Для образования глютатиона необходима аминокислота цистеин, при работе глутатионовых пероксидаз он превращается в дисульфид.
Другая группа веществ, используемых клетками нашего организма для защиты от окислительного стресса — это витамины. Реактивирует глутатиондисульфид фермент глутатионредуктаза, зависимый от НАДФН и аскорбиновой кислоты. Следовательно, для восстановления и реактивации глютатиона из [191] дисульфида нужны не только микроэлементы, но и витамины РР и С, а, по некоторым сведениям — также Е и В2 Однако, аскорбиновая кислота амбивалентна в окислительно-восстановительных реакциях и способна оказать и прооксидантные эффекты, в частности, ускоряя восстановление железа, усиливающего свободнорадикальные процессы. По Хорнсби и Кривелло, прооксидантные эффекты витамина С преобладают при его малых дозах, а антиоксидантные — при больших, что, в какой-то степени, оправдывает рекомендации Л.Полинга замедлять старение и предупреждать повреждение клеток, в частности, при вирусных инфекциях и атеросклерозе, мегадозами аскорбиновой кислоты (1983). Интересно, что, по мнению Б.Н.Эймса и соавторов (1981), у приматов таким же, как витамин С, амбивалентным действием на редокс-состояние клеток обладает мочевая кислота (см. также выше «Диатезы»).
|
|
|
Особенно тесная взаимозависимость существует между селеном и витамином Е, которые оба служат для инактивации липоперекисей. Витамин Е является сильнейшим антиоксидантом, так как ловит свободный электрон и не участвует в дальнейшей цепи. Протективное действие токоферола особенно выражено в отношении клеточных мембран. Классические эксперименты Дж.Блэнда с человеческими эритроцитами показали, что 10-дневный прием 600 ME α-токоферола ежесуточно делает плазматические мембраны 95% красных кровяных телец испытуемых резистентными к тому окислительному стрессу, который до курса лечения вызывал стопроцентный гемолиз.
Активность токоферолов восстанавливается витамином С, как и активность системы глютатиона.
Таким образом, в системе глутатиона взаимодействуют витамины, микроэлементы и серосодержащие аминокислоты. Упомянутые витамины и микроэлементы, а также полифенолы (биофлавоноиды), б-липоевая кислота и в-каротин действуют в комплексе и составляют антиокислительный резерв клеток, определяющий их резистентность к свободно-радикальному повреждению.
Многие пищевые продукты содержат значительные количества этих ингредиентов и способны насыщать ими организм.
Поэтому, питание, оптимально обеспечивающее потребности клеток в антиоксидантах, является в настоящее время предметом наиболее интенсивных разработок в диетологии.
Аутоокисление липопротеидов является одним из механизмов атерогенеза, а свободнорадикальное повреждение ДНК способно вызывать канцерогенез, поэтому антиоксиданты проявляют протективное действие в отношении атеросклероза и опухолевых заболеваний.
Дефицит незаменимых соединений, относящихся к этой системе, связан с развитием многих заболеваний, сопровождаемых клеточным окислительным повреждением.
|
|
|
Так, область, эндемичная по геохимическому дефициту селена в провинции Жэньсу (КНР) отличается повышенной частотой особой миокардиодистрофии (болезнь Кишэн), а Восточная Финляндия имеет самое низкое в мире геохимическое содержание селена и рекордно малую продолжительность предстоящей жизни сорокалетних мужчин.
Американский автор Уоллак обнаружил связь между муковисцидозом и дефицитом селена у беременных и новорожденных.
Тиоловыми антиоксидантами служат и богатые сульфгидрильными группами цитозольные белки (тиоредоксин), а также сывороточные белки макрофагального происхождения, такие как церулоплазмин, С-реактивный белок, гаптоглобин, α2-микроглобулин, амилоид А. Перечисленные белки синтезируются макрофагами различной локализации в ответ на интерлейкин-1 и ряд других медиаторов воспаления. Таким образом, осуществляется попытка предохранить организм хозяина от негативных последствий собственного окислительного удара, наносимого по агентам, вызвавшим воспаление.
Именно вследствие усиления синтеза сывороточных глобулинов — антиоксидантов происходит хорошо известное каждому медику [192] увеличение скорости реакции оседания эритроцитов (СОЭ). В связи с универсальным характером рассматриваемых механизмов, СОЭ ускоряется при широком круге инфекций, воспалений и иммунопатологических процессов разной этиологии.
Белки, усиленно синтезируемые при воспалении объединяют под названием «положительные глобулины острой фазы» (см. ниже разделы «патофизиология Воспаления» и«Преиммунный ответ»). Типичный представитель этой группы протеинов — церулоплазмин является феррооксидазой и окисляет двухвалентное железо до трехвалентного без образования свободных радикалов.
Особую роль в антиоксидантной защите играет трансферрин — отрицательный глобулин острой фазы, содержание которого в крови при воспалениях и инфекциях снижается.
Он захватывает трехвалентное железо и может переносить его в клетки.
Как уже отмечалось выше, адаптивная ценность гипоферремии при остром ответе на инфекцию экспериментально доказана и связана с каталитической ролью двухвалентного железа в системе генерации АКР.
|
|
|
Эта форма железа чрезвычайно цитотоксична, поскольку участвует в реакции Фентона и в реакциях разветвления цепей окисления мембранных липидов. При лихорадке изменения в синтезе глобулинов острой фазы ограничивают аутоокислительный эффект железа.
Кора надпочечников обладает очень высокой активностью цитохром-Р-450-зависимых оксидаз, и поэтому продукция АКР в этих органах крайне активна, особенно, в условиях стресса. Именно поэтому надпочечник обладает исключительно высокими потенциями антиоксидантных механизмов (рекордные количества аскорбиновой кислоты и витамина Е, очень высокий уровень супероксиддисмутазы). Хорнсби и Кривелло считают редокс-состояние адренокортикоцитов важным фактором, влияющим на интенсивность стероидогенеза и на морфогенез коры надпочечников, в частности, на апоптотическую гибель и эскалаторное перемещение клеток из зоны в зону, а также на смену паттерна синтезируемых клетками различной локализации кортикоидов. Следовательно, АКР влияют не только на клеточные процессы и тканевой ответ на повреждение — воспаление. Они существенны и для развития системных ответов на повреждение — реакции острой фазы и стресса (см. ниже соответствующие разделы).
Итак, свободные радикалы всегда играют определенную роль при клеточной гибели. В ряде случаев, особенно, при радиационной травме, отравлениях хлорорганическими соединениями и воспалении их вклад в механизмы повреждения клеток является определяющим. Окислительно-восстановительный баланс клеток зависит от экспрессии генов и поставки субстратов и незаменимых компонентов диеты и представляет собой одну из главных неспецифических составляющих клеточной реактивности (рис.38).
МЕХАНИЗМЫ АПОПТОЗА
Явления запрограммированной клеточной гибели известны уже более 100 лет (В.Флеминг, 1885), но оставались «в тени» некробиотических процессов, которые на протяжении десятилетий изучались намного более активно, чем программируемая гибель.
Этот вид клеточной гибели представляет собой важнейший интегральный компонент эмбриогенеза, морфогенеза и роста тканей, а также гормонозависимой инволюции. Он, наряду с лизосомальной аутофагией (см. выше) участвует в механизмах таких клеточных адаптации, как атрофия (уменьшение размеров клеток и числа функционирующих структур в них при сохранении жизнеспособности клетки) и гипоплазия (уменьшение органа вследствие уменьшения числа клеток в нем при сохранении его жизнеспособности).
|
|
|
Так например, одним из авторов этой книги было показано, что инволютивные изменения в коре надпочечников после гипофизэктомии тормозятся актиномицином Д, а значит, представляют собой активный процесс реализации [193] некой программы саморазборки клеток (А.Ш.Зайчик, 1978).
Для обозначения процесса запрограммированной клеточной гибели, морфологически и патохимически отличного от некробиоза, Н.Уолкер в 1968 году предложил термин «апоптоз». Основатели учения об апоптозе, в частности, Дж.Керр и соавт., считали понятия «запрограммированная клеточная гибель» и «апоптоз» равнозначными. В последнее время имеется тенденция применять первый термин к процессам устранения клеток в раннем онтогенезе, а понятие апоптоз относить только к программируемой гибели зрелых дифференцированных клеток. Так, указывают на наличие аутофагии и отсутствие разрывов ДНК при эмбриональной клеточной гибели, в отличие от апоптоза зрелых клеток.
Вопрос о соотношении некробиоза и апоптоза и о приуроченности этих механизмов к естественной либо насильственной гибели клеток нуждается в обсуждении.
Было бы упрощением сказать, что апоптоз это, исключительно, процесс естественной гибели клеток, а некробиоз — насильственной. Деление на эти два процесса далеко не абсолютно. Выше, обсуждая паттерны некробиоза, мы уже много раз вынуждены были упоминать об апоптозе, так как между этими процессами много общего. Дело в том, что в ответ на минимальное повреждение или повреждение, не вызывающее быстрого развития глубокой гипоксии и выраженного энергодефицита, клетки могут включать специальную программу самоуничтожения и реагировать апоптозом. В этом случае, например, при действии ионизирующего излучения или вируса СПИДа, смерть клетки насильственна, но механизм ее не некробиотический, а апоптотический. Тельца Каунсильмена обнаруживаемые при вирусном гепатите в печени, представляют собой результат апоптоза гепатоцитов под воздействием вирус-индуцированного повреждения. Это — также насильственная гибель, но механизм ее не связан с быстропрогрессирующей гипоксией и позволяет клетке успеть включить программу саморазборки. Не подлежит сомнению насильственный характер гибели клеток-мишеней под воздействием фактора некроза опухолей. Однако, несмотря на свое категоричное название, данный биорегулятор вызывает в таргетных клетках не только некроз, но и апоптоз. При реализации некробиоза и апоптоза функционируют многие общие механизмы, например, увеличение цитоплазматической концентрации ионизированного кальция и образование свободных активных кислородных радикалов. Более того, при большей силе [194] Рис.39. Механизмы апоптоза. [195] и интенсивности действия апоптогенный стимул может вызвать некробиоз, очевидно, вследствие того, что прогрессирующий энергодефицит не дает возможности клеткам реализовать энергетически «дорогую» динамику апоптоза.
Авторам представляется принципиально важным, что апоптоз, в противоположность некробиозу, это разновидность клеточной гибели, если можно так выразиться, без сопутствующего скандала.
Если некробиоз всегда сопровождается освобождением в окружающую ткань, а при массивном поражении — и в системный кровоток, медиаторов воспаления, в частности, липидных продуктов деструкции клеточных мембран, то апоптоз протекает без лейкоцитарной демаркации и перифокального воспаления, так как его механизм позволяет избежать значительного выделения медиаторов клеточного повреждения. Издание в 1996 году всеобъемлющей монографии, посвященной апоптозу («Программированная клеточная гибель» под ред. профессора В.С.Новикова) облегчает нашу задачу и делает возможным охарактеризовать в данной книге лишь наиболее общие и патофизиологически важные аспекты этой проблемы.
В.С.Новиков и соавторы выделяют следующие основные процессы, при которых доминирует гибель по типу апоптоза:
•Устранение клеток в раннем онтогенезе
•Физиологическая инволюция и уравновешивание митозов в зрелых тканях и клеточных популяциях
•Реализация процессов атрофии и регрессия гиперплазии
•Альтруистический суицид мутантных и пораженных вирусами клеток
•Клеточная гибель после слабого воздействия агентов, вызывающих при массированном поражении некроз.
Чтобы более наглядно представить отличия некробиоза и апоптоза, авторы предлагают подробно изучить таблицу 5, приводимую ими на с.193.
Важно отметить, что некроз происходит после насильственной гибели клетки в результате каких-либо причин, вызывающих глубокую тканевую гипоксию и всегда содержит литический компонент в виде либо лизосомального аутолиза, либо гетеролиза, вызываемого гидролазами фагоцитов. По современным представлениям, аутолиз при гибели клетки носит посмертный характер, а не является элементом некробиоза.
Тем не менее, раннее и значительное повреждение клеточных мембран — неотъемлемая часть процессов некробиоза, и, практически, не наблюдается при апоптозе.
Апоптоз — генетически управляемый процесс, который может быть включен различными пусковыми сигналами без какого — либо существенного предварительного повреждения исполнительного аппарата клетки, хотя может и включиться после умеренного повреждения как альтруистическое самоубийство. Устранение клеток без повреждения возможно и при экспрессии антигена стареющих клеток (см. выше — о морфогенетической роли аутофагоцитоза). Возможно, что эти механизмы «ухода без скандала» комбинируются и\или взаимодействуют.
Принципиально важно, что при неспособности вступить в апоптоз возникает неограниченно пролиферирующий клон клеток, что ведет к серьезным нарушениям в многоклеточном организме и наблюдается, например, при онкологических заболеваниях. До сих пор в данной книге мы часто упоминали об относительной полезности и потенциальной патогенности различных запрограммированных защитных процессов и приводили примеры такой «вредной полезности» или по Э.Геккелю — «опасного приспособления». В данном случае мы видим основное противоречие патофизиологии, как бы, в обратном ракурсе. Иными словами, апоптоз в клеточном цикле выступает как минимальное запрограммированное зло и также иллюстрирует основное положение наших рассуждений, так как является приспособительной смертью, гибелью по программе и своего рода «полезным вредом» [196] в чистом виде. В любом случае, наблюдения за злокачественными клетками, утратившими под действием онкогенов способность к апоптозу, доказывают, что для клеток утрата способности вовремя умереть — большое зло.
Апоптоз может начаться как ответ генов, программирующих клеточную саморазборку, на рецепторно-опосредованный сигнал (например, при стимуляции соответствующими биорегуляторами рецепторов ФНОα или глюкокортикоидного рецептора лимфоцитов).
Не только ФНО и глюкокортикоиды, но и почти все цитокины, включая 13 интерлкейкинов и 3 интерферона могут быть кодовыми сигналами апоптоза, причем в одних клетках они его запускают, а в других — ингибируют. Тканеспецифические факторы роста и гемопоэтины являются ингибиторами апоптоза для своих клеток-мишеней. Тропные гормоны гипофиза оказывают свой трофический эффект на железы-мишени также путем ингибирования апоптоза.
Сигнал может оказывать на клетку разнонаправленное в отношении апоптоза действие, в зависимости от исходного состояния мишени, как это описано выше для ФНО.
В роли генетических индукторов апоптоза, срабатывающих в ответ на рецепторный сигнал, могут выступать гены Fas/APO-1, c-мус, мах, р53, ced-З и другие. Подавление экспрессии некоторых генов, например, вс1 -2, также вызывает апоптоз. Детальное изучение механизмов, с помощью которых продукты этих генов запускают или сдерживают апоптоз, только начато. Однако, уже выяснено, что они могут усиливать образование активных кислородных радикалов (как белок АРО-1, гомологичный рецептору фактора некроза опухолей) регулировать перенос кальция в цитоплазму (как продукт гена вс1 -2), запускать нейтральные протеазы цитозоля (как продукт гена ced-З), связываться с ДНК (как димер белков мус-мах).
Принципиально важно, что апоптоз может быть индуцирован даже в безъядерных постклеточных структурах. Следовательно, первичным звеном апоптоза могут быть не только ядерные события, но и определенные метаболические изменения в цитоплазме или активация долгоживущих матричных РНК, как в случае с антигеном стареющих клеток.
Г.А.Софронов и соавторы полагают, что инициировать апоптоз могут активные кислородные радикалы (АКР). При умеренных повреждениях клетки в отсутствие гипоксии происходит редукция трансмембранного потенциала митохондрий и генерация ими АКР. Если антиоксидантные системы клетки не компенсируют сдвига редокс-потенциала, процесс прогрессирует. При условии отсутствия выраженного энергодефицита и сохранности генетического аппарата реализуется апоптоз, но глубокая гипоксия и выраженные повреждения ДНК инициируют некробиоз. При развитии апоптоза АКР изменяют условия взаимодействия кальция с кальмодулином и способствуют нарастанию цитоплазматической и внутриядерной активности (а при блокаде гена вс1-2 — и росту внутриклеточной концентрации) кальция.
Кальций-зависимое звено механизма апоптоза активирует кальпаины, что ведет к протеолизу белков цитоскелета, образованию цитоплазматических выпячиваний, разрушению межнуклеосомных связей в ядре.
Активируется кальцийзависимая эндонуклеаза. Это провоцирует упорядоченные межнуклеосомные разрывы хроматина и фрагментацию ядра. Кальций-зависимая трансглютаминаза агрегирует цитозольные белки. Конечным этапом процесса служит распад клетки на апоптотические тельца и их аутофагоцитоз (см. рис.39)
Основные патохимические механизмы апоптоза частично охарактеризованы также в предыдущих разделах книги, при сравнении с некробиозом.
следующая глава
предыдущая глава
содержание
|
|
|
