 |


Глава шестая. Определение фазового. И элементного состава порошков. 6. 1. Методы фазового анализа
|
|
|
|
Глава шестая
ОПРЕДЕЛЕНИЕ ФАЗОВОГО
И ЭЛЕМЕНТНОГО СОСТАВА ПОРОШКОВ
Диагностика порошков включает определение элементного ифазового состава в качестве одной из основных их характеристик, поскольку материал порошка и его кристаллическое строение в решающей степени определяют физико-химические имеханические свойства самих порошков и полученных из них изделий. Совреvенные методы фазового и химического анализа позволяют определять фазовый и элементный состав порошка в целом и отдельных его частиц [1-3], а также состав и структуру поверхностных слоев [4, 5], составляющих значительную часть объема порошков, особенно высокодисперсных.
6. 1. Методы фазового анализа
Основную группу методов фазового анализа составляют дифракционные методы: рентгеновский, электронографический и нейтронографический [6], которые применяются для изучения фазового состава порошка в целом. Фазовый состав отдельных частиц порошка определяется с помощью локальных методов исследования, некоторые из которых также будут здесь рассмотрены.
Рептгепографический фазовый анализ. Рентгеновский фазовый анализ, как и другие дифракционные методы, основан на том, что каждое кристаллическое вещество дает специфическую дифракционную картину с определенным количеством, расположнием и интенсивностью линий [7 -15]. Дифракционная картина фотографируется на рентгеновскую пленку в камерах Дебая (РНД, РНУ) или фиксируется с помощью дифрактометров ДРОН любой модификации.
Применение дифрактометров повышает точность, чувствительность иэкспрессность исследований [7 -8]. Фотометод дает дополнительные по сравнению с дифрактометрическим, сведения для разделения линий, принадлежащих фазам с разной дисперсностью ипредысторией, что облегчает проведение фазового анализа порошков с гетерогенной структурой и порошковых смесей [9, 10]. Основой для такого разделения является вид линий: сплошные или точечные, резкие или размытые. В менее сложных случаях фазовый анализ следует проводить на дифрактометре и по возможности отбирать для съемки порошки размером около микрона, так как порошки такой величины дают самые узкие гладкие пики.
|
|
|
Рентгенографически могут быть исследованы и более мелкие порошки, но, начиная с размера около 1000 А, они будут давать, сильно размытые линии [12-15]. Порошки, крупнее 10-50 мкм, если только они не состоят из более мелких зерен, будут давать зубчатый профиль пика на дифрактограмме, а потому образцы и них должны обязательно вращаться во время съемки. Хрупкие порошки можно измельчить до нужного размера растиранием в ступке. Растирать порошки из шrастичных металлов не рекомендуют, так как это приводит к размытию линий.
Для изготовления образца для съемки на дифрактометре следует добавить в порошок минимальное количество клея, не дающего своей дифракционной картины, ипоместить его в кювету таким образом, чтобы поверхность образца была ровной исовпадала с верхним краев кюветы. Не рекомендуется запрессовывать порошок в кювету даше сравнительно легким нажимом, так как это может создать текстуру в образце и сильно изменить относительную интенсивность линии. Для предотвращения образования текстуры в образце, особенно при исследовании сильно неравноосных частиц, к снимаемому порошку добавляют некоторое коли чество слабопоглощающего вещества (мука, крахмал), которое, обволакивая частицы порошка, помогает им располагаться беспорядочно [11].
Указанные требования к образцам для съемки особенно существенны при проведении количественного фазового анализа. Если качественно фазовый состав смеси известен, то по относительной интенсивности линий разных фаз можно оценить их количественное соотношение (метод гомологических пар), так как интенсивность линий каждой из фаз пропорциональна количеству ее в смеси. В другом варианте для проведения количественного фазового анализа измеряют интенсивности линииисследуемого образца иэталона (метод подмешивания эталона иметод независимого эталона) [16, 17].
|
|
|
Минимальное количество порошка, необходимое для анализа, можeт составлять миллиграммы, так как для приготовления образца можно пользоваться не кюветой, а подложкой из оргстекла, на которую с помощью рамзальской замазки порошок наносят очень тонким слоем. Труднее указать минимальное процентное содержание фазы в порошке, которое может быть обнаружено рентгенографическим методом. Оно меняется от 1 до 10% и зависит как от химического состава пробы и симметрии кристаллической решетки определяемой фазы, так и от дисперсности этой фазы [14, 15].
Чувствительность метода можно повысить, особым образом препарируя образцы. Если порошки содержат малое количество мелкодисперсных включений, например карбидов, нитридов, интерметаллидов и т. д., распределенных в большом количестве другой фазы, то путем химического или электрохимического травления можно выделить частицы изучаемой фазы (см. с. 191) и исследовать их отдельно. Разновидностью способа является неполное
вытравливание, при котором поверхность таблетки, спрессованной из порошка, обогащается нерастворившейся фазой.
Результаты количественного и качественного анализа металлических порошков могут быть искажены за счет особенностей строения частиц гетерофазных порошков [19]. В большинстве случаев при поверхностном взаимодействии металлических порошков с газами и напыленными элементами наблюдается послойное расположение фаз в частицах. Так как рентгеновская дифракционная картина образуется от поверхностного слоя небольшой глубины (30-50 мкм), то для порошков указанного строения рентгеновские линии металлического ядра будут ослаблены из-за поглощения лучей в наружном слое либо полностью отсутствовать. В результате этого создается ложное впечатление об отсутствии металлической фазы в порошке или будет неправильно оценено количественное соотношение фаз в пробе. Чтобы получить дифракционную картину от внутренних слоев исследуемых частиц, необходимо использовать жесткое рентгеновское излучение или перед съемкой растирать содержащие хрупкую фазу порошки [18]. Изменения фазового состава порошков обнаружены при хранении в результате взаимодействия с элементами окружающей атмосферы [19], при плазменной переплавке и получении порошков распылением под влиянием распыляющей и охлаждающей среды.
|
|
|
В процессе получения порошков плазменным и газовым распылением фазовый состав исходного материала может измениться также за счет фиксирования высокотемпературной кристаллической модификации или пересыщенного твердого раствора взазен равновесной гетерогенной структуры [20-23]. Обнаружены отличия в фазовом составе различных по крупности фракций порошков, полученных распылением двух и многокомпонентных сплавов. Электронографический анализ. Электронография является электронным аналогом рентгенографии [24]. Волновые свойства электронов позволяют получать дифракционную картину от кристаллической решетки, подобную рентгенографической. Основной является разница в длинах волн используемого излучения: рентгенография около 1 А, электронография - 0, 05-0, 07 А. Прямым следствием малой длины волны электронных пучков является увеличение разрешающей способности для малых областей рассеяния, благодаря чему появляется возможность проводить фазовый анализ ультрадисперсных порошков. При анализе порошков
с величиной частиц менее 1000 А рентгеновские линии начинают
уширяться (размываться) и при размере частиц около 200 А становятся практически неразличимыми. Электронография дает ддя этого размера кристаллов очень резкие линии, а уширение мало даже в том случае, когда размер частиц или кристаллитов в частицах составляет 100 А. Таким образом, дифракция электронов на малых частицах дает более надежные данные для фазового анализа, чем дифракция рентгеновских лучей.
|
|
|
Электронографические исследования проводят с помощью специальных
приборов - электронографов, исполненных в вертикальном или горизонтальном варианте. Наиболее совреременный вертикальный прибор - ЭГ-100 А. Можно также использовать почти любой электронный микроскоп после соответствующего изменения режима его работы. Исследуемый образец облучают в вакууме пучком электронов сечением несколько мм2, и образующуюся дифракционную картину наблюдают на флуоресцирующем экране или фотографируют на пленку. Метод обладает высокой чувствительностью, и для анализа достаточно иметь 10-5 г исследуемого вещества.
В современных электронных микроскопах имеется приспособление, дающее возможность получать электронограммы от малого участка образца (0, 5-2 мкм), т. е. осуществлять так называе rую микродифракцию. Совместное проведение электронографических и электронно-микроскопических исследований дает сведения как о внешней форме частиц, так и их фазовом составе.
Существует два метода съемки электронограмм: на просвет и на отражение. В первом случае используют образец толщиной до нескольких сот ангстрем, во втором - плоский шлиф, установленный так, что электронный луч практически скользит по его поверхности [25]. Методом на проснет изучают изолиронанные тонкие пленки иультрадисперсные порошки. Для анализа порошков берут обычно каплю взвеси порошка в спирте (или в воде) инаносят ее на специально приготовленную тончайшую пленку из 1%-ного раствора коллодия [11]. Методом на отражение изучают состояние поверхностных слоев, фазовый состав поверхностных пленок, а также ультрадисперсных частиц, распределенных в матрице. В последнем случае подбирают такие режимы химического или электрохимического травления, чтобы матрица растворялась сильнее, а исследуемые частицы образовывали выступы или даже скопления частиц в виде осадка на поверхности шлифа [11]. Скользя по этим мельчайшим выступам, электронный пучок образует нх дифракционную картину.
Методом электронографии решаются задачи, связанные с изучением «малых количеств» высокодисперсных порошков, а также весьма тонких изолированных пленок. На ультрадисперсных порошках удается обнаруаашать тончайшие слои окислов, нитридов и т. д., образующихся при взаимодействии их с газами. При съемке на отражение можно определять фазовый состав высокодисперсных включений в стали и сплавах, выделений второй фазы при распаде пересыщенных твердых растворов, включений, находящихся на поверхности межзеренного хрупкого излома, оксидных и других пленок.
|
|
|
С помощью электронографии может быть проведено определение параметра кристаллической решетки (см. с. 189) и, хотя из измерения производятся со значительно меньшей точностью, чем по рентгенограммам, этот недостаток искупается возможностью получения указанной характеристики для ультрадисперсных порошков.
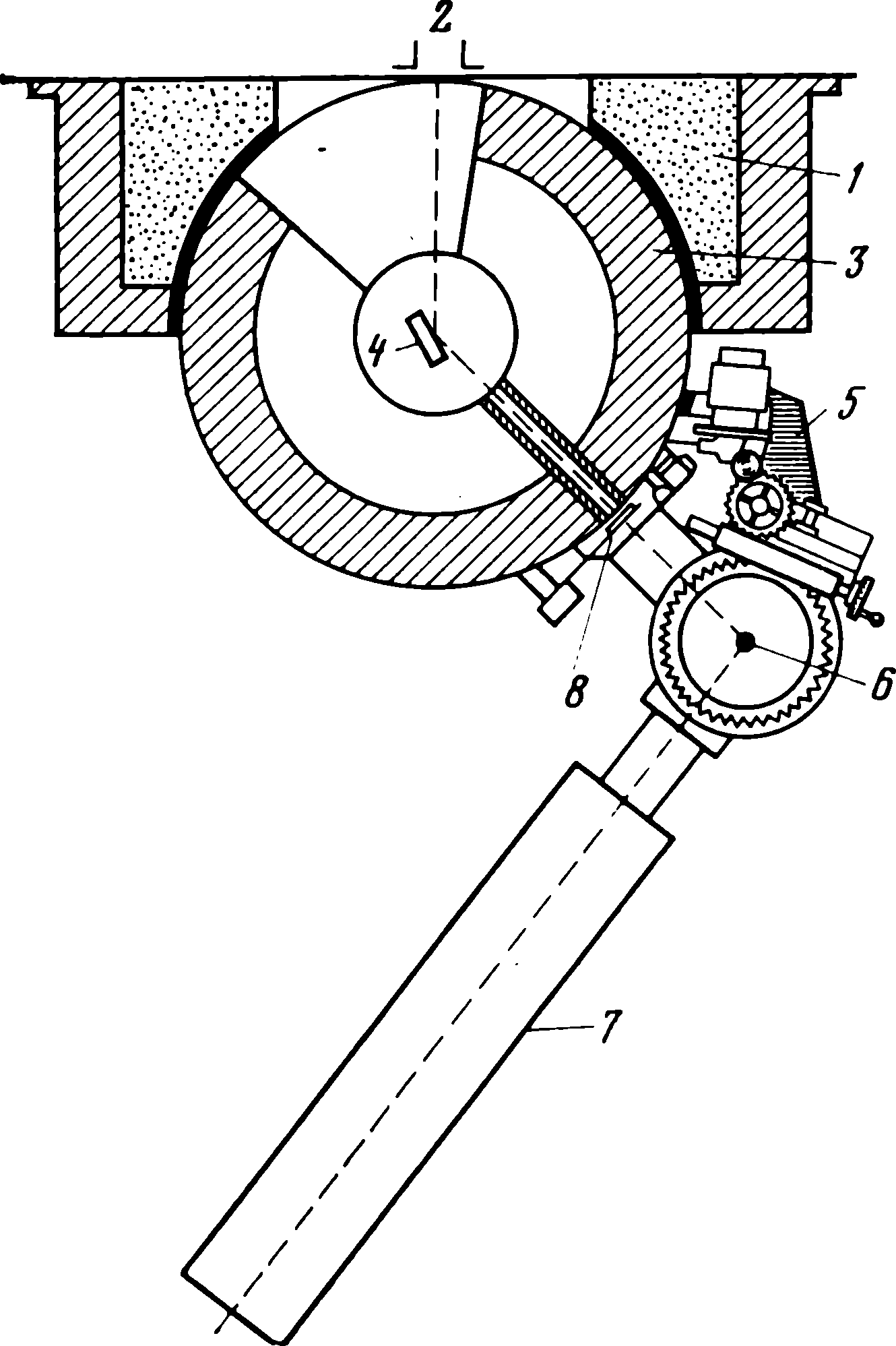 Нейтронографический анализ. Нейтроны рассеиваются веществом слабее, чем рентгеновские лучи; амплитуды рассеяния нейтронов, особенно для элементов с z> 10, существенно меньше, чем для рентгеновских лучей. Для легких элементов они одного порядка. Образцы для нейтронографических исследований в связи с малой амплитудой рассеяния нейтронов должны и иметь размер порядка миллиметров. Характерно, что нейтроны одинаково рассеиваются под всеми углами θ и строгая зависимость рассеяния от атомного номера отсутствует.
Нейтронографический анализ. Нейтроны рассеиваются веществом слабее, чем рентгеновские лучи; амплитуды рассеяния нейтронов, особенно для элементов с z> 10, существенно меньше, чем для рентгеновских лучей. Для легких элементов они одного порядка. Образцы для нейтронографических исследований в связи с малой амплитудой рассеяния нейтронов должны и иметь размер порядка миллиметров. Характерно, что нейтроны одинаково рассеиваются под всеми углами θ и строгая зависимость рассеяния от атомного номера отсутствует.
Длина волны в ангстремах, присущая тепловым нейтрона, обычно применяемым для структурных исследований, λ
Схема нейтронного спектрометра приведена на рис. 6. 1 [25].
Пучок нейтронов вырезается коллиматором 2 длиной 50-150 см. Пучок падает на монохроматор 4, установленный под брэгговским углом (для данной длины волны нейтронов). Для монохроматизации пучка обычно используют монокристаллы свинца или меди, а также пиролитического графита, имеющего высокую отражательную способность.
Монохроматор окружен защитой из парафина 1 и свинца 3. С его помощью из сплошного спектра вырезается пучок с интервалом длин волн 0, 15 А, что на два порядка превышает собственную ширину линии рентгеновского спектра. Большая спектральная ширина монохроматизированного пучка нейтронов не позволянет определять периоды решетки с погрешностью меньшей 0, 1 А. Монохроматизированный пучок нейтронов попадает на образец 6, установленный на гониометре 5 нейтронного спектрометра. Пучок нейтронов можно перекрывать кадмиевой заслонкой 8. Рассеянные образцом нейтроны регистрируются газоразрядным счетчиком 7.
Обычно нейтронографические исследования проводят в сочетаниис рентгено- и электронографическими. Использование тепловых нейтронов позволяет иметь данные, которые не могут быть получены или получаются с плохим разрешение с помощью рентгено- или электронографии.
Особенности ядерного рассеяния нейтронов используют при исследовании структуры соединений из элементов с близкими атомньши номерами, в частности при изучении явлений упорядочения, сопровождающихся изменением взаимного расположения атомов с близкими атомными номерами (например, система Fe-Co) [26].
При изучении ультрадисперсных порошков по соотношению пиков и диффузного фона может быть сделан вывод о соотношении размеров областей с неискаженной решеткой и решеткой искаженной атомами, внедренными через поверхность из окружающей среды в процессе получения порошков иих хранения [27].
Определепие параметра кристаллической решетки. По значе ниям межплоскостных расстояний d, определяемых для данного вещества при проведении фазового анализа, можно определить параметры элементарной ячейки данного вещества. Большинство металлов имеют кубическую структуру, и параметр решетки для них можно рассчитывать по формуле:
 (6. 1)
(6. 1)
где h, k, l - индексы отражения. Для материалов с некубической структурой применяются иные формулы. С учетом уравнения
Вульфа - Брегга 2dsinθ = nλ (6. 1) получаем
(6. 2)
где λ - длина волны используемого излучения; θ - угол отражения.
Прирешении некоторых задач, таких, как определение концентрации твердых растворов, границ растворимости ит. п., точность определения периода решетки методами, применяемыми в фазовом анализе, оказывается недостаточной. В этпх случаях измерение периодов решетки проводится с помощью более точных, прецизионных методов [10, 14, 28].
Относительная погрешность определения межплоскостного расстояния уменьшается с увеличением угла отражения по формуле ∆ d/d = ctgθ ∆ θ (6. 3)
и прецизионности в определении параметра решетки добиваются в первую очередь увеличением угла отражения, по которому ведется расчет, что достигается соответствующим выбором излучения. При фотографической регистрации параметр определяют для нескольких линий и проводят аналитическую или графичесиую экстраполяцию [10]. При работе на дифрактометрах проводят прецизионное определение угла θ по профилю линии [7] и вводят соответствующие поправии [14]. Применяются также специальные математичесюrе методы обработки результатов [29-33]. Способы определения параметров решетки по рентгенонейтрона- и электро н ографическим данным во многом аналогичны.
Измерения параметра решетки широко применяются при исследовании порошков. В процессе получения легированных металлических порошков методом распыления расплава происходит образование пересыщенных твердых растворов. Когда скорость охлаждения достаточно велика, концентрация легирующих элементов в твердом растворе может в десятки раз превышать концентрацию этих элементов в равновесном состоянии [21]. Если атомы легирующих элементов отличаются по размерам от атомов матрицы, то с помощью прецизионного измерения параметра решетки твердого раствора можно определить концентрацию легирующих элементов в твердом растворе [34].
Представляет интерес также определение параметра кристаллической решетки ультрадисперсных порошков. Свойства ультрадисперсных порошков существенно отличаются от свойств традиционных материалов, применяемых в порошковой металлургии. Отличие это в значительной степени связано со спецификой структуры мелких частиц (размерами 1000 А и менее), из которых состоят ультрадисперсные порошки. Рентгенографические исследования [27] показали, что параметры кристаллической решетки частиц магния размерами 295 А и кадмия размерами 650 А заметно меньше их значений в поликристаллических образцах.
С помощью нейтронографии изучалась структура ультрадисперспых порошков никеля, олова, алюминия и меди [27]. Нейтронографический метод исследования ультрадисперсных порошков имеет преимущества перед рентгено- и электронографией, поскольку из-за ядерной природы амплитуда когерентного рассеяния нейтронов не зависит от угла. Измерения проводились с помощью потока мопохроматизированных нейтронов с длиной волны 1, 078 А. Обпаружено уменьшение параметра кристаллической решетки всех исследуемых порошков по сравнению с литым металлом. Параметр кристаллической решетки ультрадисперсного алюминия с величиной частиц 400 А после его приготовления равен 4, 02±0, 1 А, что меньше, чем для обычного алюминия (4, 0496 А).
Однако нейтронограмма, снятая через 1, 5 года с этого же образца алюминия, показала, что структура претерпела изменения, хотя и осталась кубической. По-видимому, образовался окисел алюминия за счет поглощения кислорода, но он не идентифицировался ни с одним известным окислом алюминия.
Вообще малые частицы ультрадисперсных порошков легко взаимодействуют с кислородом из окружающей среды из-за большой активности. Внедряясь с поверхности, кислород может образовывать слои твердых растворов, окислов или разупорядоченных фаз, уменьшая размеры областей когерентного рассеяния исходных металлов. Под воздействием поверхностных слоев с измененными межатомными расстояниями, а также в связи с влиянием лапласовского давления происходит перестройка структуры и внутренних слоев. При этом уменьшаются параметры кристаллической решетки и увеличиваются смещения атомов из своих идеальных положений, характерных для массивных образцов [27].
Прочие методы фазового апализа. Изучение фазового состава малого количества неметаллических и интерметаллических фаз в металлических порошках проводят металлографическим, петрографическим и химическим способом, причем первые два осуществляют с использованием тех же шлифов, что идля изучения микроструктуры. Селективное химическое растворение порошков не представляет трудностей, но более перспективным является метод селективного электролитичес: кого разделения фаз, который заключается в анодном растворении металлического материала сложного состава. При этом в зависимости от режима растворения и состава электролита нерастворенной оказывается та или иная неметаллическая фаза. Полученный анодный осадок подвергается дальнейшему исследованию микрохимическим, рентгенографическим и другими методами.
Приемлемость электролитического способа для фазового анализа порошков имеханизм растворения порошковых материалов изучались в [35-37]. Учитывая пористость ималую прочность спрессованных (неспеченных) образцов, для выделения карбидной иоксидной фаз из железных порошков [35, 36] применялись электролиты, отличные по своему составу от электролитов, используемых при исследовании литых материалов того же состава.
Для фазового анализа отдельных порошковых частиц или включений в порошках используются методы рентгеновского (РМ), электронного (ЭОС) иионного (БИМС) микроанализов. Ниже указаны способы определения фазового состава поверхностных слоев (ЭСХА идр. ) и возможность проведения фазового анализа с помощью эффекта Мессбауэра.
|
|
|
