 |


Глава седьмая
|
|
|
|
195
анализа по флуоресцентному (вторичному) излучению исследуемый образец помещают вблизи анода мощной рентгеновской трубки. Первичное излучение, выходящее из трубки, возбуждает вторичное характеристическое излучение исследуемого вещества. Это излучение выделяется в параллельный пучок с помощью щели Соллера и попадает на кристалл, который paзлaгaeт его в спектр. Спектр регистрируется с помощью газоразрядных или сцинтилляционных счетчиков.
Поскольку исследуемый образец находится вне рентгеновской трубки, на проведение анализа затрачивается времени не больше, чем при исследовании методом оптического анализа. Этот метод анализа обладает очень высокой чувствительностыо, равной 0, 04-0, 0005 %.
Флуоресцентный рентгеноспектральный анализ имеет некоторые преимущества по сравнению с анализом по первичным спектрам: 1) возможность быстрой смены образца, так как образец располагается на воздухе, вне объема рентгеновской трубки или вводится в него через шлюз; 2) в процессе анализа образец не нагревается, поэтому химический состав его не изменяется и возможно исследование даже легкоиспаряющихся веществ; 3) применение метода внешнего стандарта позволяет получить высокую точность;
1) во вторичном спектре отсутствует фон непрерывного спектра,
что приводит к повышению контрастности аналитических линий, т. е. повышается чувствительность анализа.
К недостаткам флуоресцентного метода относятся следующие:
1) вторичные спектры имеют малую и интенсивность, поэтому не применим фотографический метод регистрацпп; 2) интенсивность аналитических линий существенно зависит от общего состава пробы, особенно при наличии мешающих элементов, т. е. элементов, влияющих своим флуоресцентным излучением на интенсивность линийстандарта или анализируемого элемента; 3) чувствительность флуоресцентного метода анализа по спектрам в 5- 7 раз ниже, чем по К-спектрам [25, 43].
|
|
|
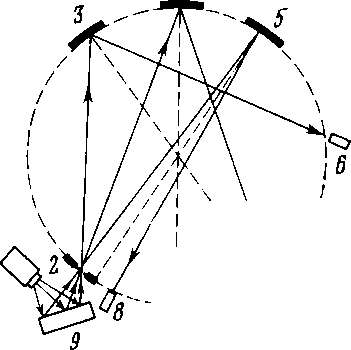
Для проведения анализа флуоресцентным методом используют многоканальные спектрометры (квантометры), которые состоят из нескольких (2, 5, 10) спектрометров, на кристаллыанализаторы которых попадает флуоресцентное излучение от исследуемого образца (рис. 6. 2). . Каждый из спектрометров настроен на спектральную линию определенного элемента, интенсивность которой измеряется с помощью электронно-счетного устройства. Прибор указывает одновременно содержание нескольких элементов. Один из каналов используется для измерения спектральной линии стандарта. Анализ на многоканальных спектрометрах требует меньших затрат времени и дает большую точность, чем на одноканальных спектрометрах.
Кроме обычного флуоресцентного метода, при котором вторичное характеристическое излучение образца разлагается в спектр с помощью кристаллов, т. е. дисперсионного метода, широкое распространение получили бескристальные (бедисперсионные) рентгенофлуоресцентные
аппараты. Эти аппараты обладают существенным выигрышем в светосиле, поскольку производят в103 больший поток вторичного излучения.. Поэтому вбескристальных аппаратах для возбуждения вторичного излучения образцов используют миниатюрные рентгеновские трубки мощностью 0. 5-10, 0 вт или радиоизотопные источники (55 F e, 1 47 Р ш,
109 Cd, 3 H/Zr).
Порог чувствительности бес- кристального анализа выше, чем
| \ |
| \ |
| \ |
| / |
| \ |
| / |
| _1 j --/ |
| 7 |
Рис 6. 2. Простейшая схема рентге новского многоканального спектро метра
у дисперсионного на 1-2 по- 1 - рентгеновсиая трубка; 2 - входная рядка. Меньше Иточность бес- щель; 3-5 - кристаллы; б-8 - счетчики кристального анализа при низ- 9 - исследуемый образец
|
|
|
ком содержании определяемого
элемента в пробе 0. 1- 0, 5 %. При содержании элемента > 1 % точность анализа на бескристальном аппарате уже близка к точности дисперсионного метода анализа.
Бескристальные анализаторы удобно использовать при серийном анализе однотипных образцов на ограниченное число элементов. Для качественного анализа неизвестных образцов или количественного определения большой группы элементов предпочтительнее дисперсионные спектрометры и квантометры.
Анализируемые порошки помещают в кювету или прессуют в таблетки; облучению подвергается значительная площадь образца. В настоящее время создаются приборы для локального микроанализа по флуоресцентным спектрам (РФМА) [3]. Этот метод обеспечивает возможность локального анализа с разрешением до 0, 1 мм и микроанализа из пробы 10-3 – 10-6 г с абсолютным пределом обнаружения до 10-10 г, что на три порядка лучше, чем при обычном флуоресцентном анализе. Для изучения порошка этим методом может быть сделан шлиф, а могут быть использованы и частицы произвольной формы без предварительной подготовки пробы, что ускоряет исследование иобеспечивает полную сохранность анализируемых объектов.
Микрорентгеноспектральпый апализ. Рентгеноспектральный
микроанализ (РСМА, МРСА) является локальным методом исследования, позволяющим проводить определение элементного состава в микрообъеме вещества [44-47].
Локальность РСМА достигается тем, что анализируемое рентгеновское излучение возбуждается в очень малом объеме - в месте бомбардировки шлифа узким пучком электронов - электронным зондом. Фокусировка электронов до диаметра 0, 1-3 мкм осуществляется с помощью электронно-оптической системы. Возбуждаемое рентгеновское характеристическое излучение
разлагается в спектр кристаллом-анализатором и фиксируется счетчиками рентгеновских квантов.
Метод обладает очень высокой локальной чувствительностью (10-13-10-14 г) и достаточной точностью. Погрешность количественного микроанализа составляет 1-10 относительных. При проведении количественного анализа используется метод внешнего стандарта, причем в качестве внешнего стандарта обычно выбирают чистые элементы или простейшие соединения. Конструкция микроанализаторов позволяет провести качественный и количественный анализ в точке (путем регистрации интенсивности каждой линии спектра), а также установить изменение концентрации данного элемента вдоль выбранного направления сканирования (путем измерения интенсивности одной линии при непрерывном перемещении-шлифа под пучком электронов).
|
|
|
Современные модификации приборов позволяют проводить анализ на элементы, начиная с (Z = 4) до (Z = 92).
Для проведения РСМА порошков необходимо, как и при исследовании монолита, приготовить полированный шлпф, для чего порошок заливают в твердеющую массу, желательно токопроводящую. В противном случае на шлиф напыляют тонкий (100-300 А) токопроводящий слой из металла или углерода. Таким способом обычно исследуют крупные порошки. Разрабатываются методы качественного и количественного анализа отдельных частиц порошка не с полированной, а с рельефной поверхностью [1, 2, 48]. Частицы порошка от одного до нескольких микрон помещают на бериллиевую полированную подложку и закрепляют на ней с помощью тонкой пленки коллодия [48]. В других вариантах на поверхность рельефных частиц, находящихся на бериллиевой подложке, дополнительно напыляют углеродную пленку [1]. Для разделения частиц друr от друrа применяют предварительное диспергирование их в растворе с помощью ультразвукового диспергатора, и каплю этого раствора наносят на полированную поверхность пластины из металлического бериллия. С целью отбора частиц определенного размера, а также для исключения очень мелких частиц применяют седиментационный анализ [1].
Рельеф поверхности и размер частиц вносят значительные изменения в интенсивность линий рентгеновского спектра и, следовательно, влияют на результаты количественных измерений. Поэтому для количественного анализа рельефных частиц разрабатываются методики учета этих факторов путем введения расчетных поправок, а также использования в качестве эталонов наряду с полированными образцами рельефных частиц, приготовленных тем же методом, что и исследуемые [1].
|
|
|
Методом РСМА изучают фазовый и химический состав отдельных микронных частиц, оценивают степень неоднородности частиц по составу [1, 48], изучают контактное взаимодействие всмеси порошков [49], распределение элементов по структурным ифазовым составляющим в средних и крупных частицах порошков и спеченных материалах [50].
Мессбауэровская спектроскопия. Ядерный гамма-резонанс (ЯГР) основан на эффекте Мессбауэра, rоторый за. нлючается в селективном взаимодействии у-квантов, испускаемых излучателем, с поглощающим веществом при условии идентичности их атомов. Эффект наблюдается лишь в том случае, когда энергия излучаемых у-квантов полностью передается ядрам атомов поглотителя. Энергию отдачи ядер атомов можно значительно уменьшить, если ядра жестко закрепить в кристаллической решетке, тогда импульс отдачи
Рис. 6. 3. Блок-схема мессбауэровского спектрометра
1 - движитель (для возвратно-поступательноrо перемещения источнина относително поглотителя); 2 - источнин у-нвантов; з - поrлотитель (исследуемый образец); счетчич у-нвантов; 5 - комплент спектрометричесной аппаратуры; 6 - комплект регистрирующей и управляющей аппаратуры
воспринимают не отдельные ядра, а вся кристаллическая решетка [51, 52].
Источником у-квантов для экспериментального наблюдения эффекта является радиоактивный изотоп, который прираспаде переходит вначале в возбужденное состояние стабильного изотопа, а затем - в основное его состояние и испускает при этом квант у-излучения. Наиболее распространенной мессбауэровской парой являются радиоактивный изотоп 57Со и стабильный изотоп 07 Fe: источником у-квантов служат атомы изотопа 07 Со, внедренные в металлическую матрицу из Cr, Cu, Pd или Pt, поглотителем у-излучения - ядра 57Fe, находящиеся в исследуемом объекте. Простейшая блок-схема мессбауэровского спектрометра приведена на рис. 6. 3.
Для наблюдения резонансного поглощения необходимо в очень небольших интервалах изменять энергию у-квантов, что достигается за счет эффекта Допплера при перемещении источника 2 относительно поглотителя 3. Чтобы пройти все возможные резонансные энергетические состояния исследуемого поглотителя, для указанной пары необходима скорость относительного перемещения ±1 см/с, что реализуется с помощью электродинамического движителя и генератора электрических колебаний.
Исследуемые объекты должны представлять собой порошки размером около 20-10 мкм и менее или фольгу. Толщина образца в пересчете на железную фольгу должна быть не более 50 мкм, оптимальная площадь объекта 1-2 см2
|
|
|
Резонансное поглощение у-квантов регистрируется в виде зависимости числа у-квантов, прошедших через поглотитель в единицу времени (N), от скорости движения v (или ∆ Е). Величина экспериментально наблюдаемого эффекта поглощения у-квантов (площадь под резонансной кривой) определяется силами связи резонансного
атома с матрицей, в которой он находится. Наибольшее воздействие на вид спектра резонансного атома оказывает его ближайшее атомное окружение, т. е. число и сорт атомов, находящихся в первой и второй координационной сфере.
Если резонансный атом будет находиться в магнитном или неоднородном электрическом поле, то это приведет к усложнению мессбауэровского спектра. Например, 57Fe в парамагнитной матрице кубического кристалла имеет спектр в виде одиночной резонансной линии. Соответствующий спектр для случал некубической матрrщы, как правило содержит две компоненты. В ферромагнитном кристалле образуется шестикомпонентный спектр. Изменение валентности резонансного атома также будет приводить к изменению спектра.
Даже то немногое, что сказано о ЯГР-спектрах, показывает, что они дают возможность проводить тонкий фазовый анализ (структурный и магнитный) порошкового объекта. Причем следует сказать, что дисперсность порошка (вплоть до размеров 50-20 А) не влияет на качество анализа.
К настоящему времени накоплен большой экспериментальный материал по ЯГР-спектрам различных объектов, что вместе с использованием ЭВМ [53] для расшифровки сложных спектров позволяет достаточно быстро проводить соответствующие исследования. Ядерный у-резонанс был использован для изучения особенностей строения и свойств ультрадисперсных порошков железа [54], никеля [55], механических и аморфных порошков сплавов [56, 57]. Широкое применение мессбауэровская спектроскопия находит в приложении к химии [58].
В настоящее время разработано большое число методов диагностики поверхности [4, 59, 60]. Многие из этих методов, позволяющих осуществлять диагностику поверхности, широко применяются для изучения порошков, как раз имеющих сильно развитую поверхность. Под поверхностью в этих исследованиях понимают, как правило, часть объема материала толщиной примерно до 10 атомных слоев. Но сведения могут быть получены и от всего объема, если наружные слои будут последовательно удаляться.
Для зондирования поверхности пригодны пучки электронов, ионов, фотонов или нейтральных атомов. Все они вызывают эмиссию вторичных частиц - электронов, ионов, фотонов или нейтралъных атомов. Поэтому разные методы анализа поверхности можно классифицировать в соответствии с видом зондирующего воздействия и типом эмитируемых частиц. Анализ последних позволяет получать информацию о природе частиц, их пространственном и энергетическом распределении и количестве.
Из всего многообразия используемых и развивающихся методов рассмотрим метод рентгеновской фотоэлектронной спектроскопии, электронную оже-спектроскопию и вторично-ионную массспектрометрию.
Метод рентгеновской фотоэлектронной спектроскопии (РФЭС) основан на получении и анализе спектров электронов, испускаtvs[
атомами вещества при поглощении фотонов рентгеновского излучения, т. е. при фотоионизации атомов. Возникающий под действием рентгеновского излучения спектр фотоэлектронов представляет собой наложение дискретных спектров рентгеновских фотоэлектронов и оже-электронов. Существует близкий метод - фотоэлектронная спектроскопия в вакуумной ультрафиолетовой области (УФЭС), - в котором источником фотонов является монохроматическое ультрафиолетовое излучение.
Как и любой метод спектрального анализа, РФЭС включает в себя изучение зависимости числа выброшенных из образца электронов от их кинетической энергии, т. е. прецизионное определение энергетического положения максимума линий и их интенсивностей. Исследования спектров проводятся с помощью магнитных или электростатических бета-спектрометров, рассчитанных на энергии 3-10 кэВ [61, 62].
Наиболее простым методом приготовления образца для съемки является использование липкой лепты. При применении порошкообразного образца следует следить за тем, чтобы связующее вещество не покрывало облучаемую поверхность образца. Образец также может быть впрессован в проволочную сетку. Исследуемые порошки могут быть также впрессованы в мягкие металлические поверхности, которые специально делаются шероховатыми путем травления [63].
Электронная спектроскопия дает возможность получать фундаментальные сведения об уровнях энергии атомов в различных соединениях. Сдвиги этих уровней при переходе от одного соединения данного элемента к другому характеризуют перераспределение электронной плотности и характер химической связи. Электронная спектроскопия позволяет получать разделяющиеся пики от одного и того же элемента различной валентности. Широкое применение этого метода для химического анализа дало ему еще одно название - электронная спектроскопия для химического анализа (ЭСХА). ·
Изучение структуры энергетических уровней атомов в различных соединениях открывает широкие возможости для практического использования электронных спектров в фазовом анализе. Методом ЭСХА без разрушения образца можно идентифицировать все химические элементы, кроме водорода [4]. Метод позволяет проводить как качественный, так и количественный анадиз.
Рентгеновское излучение трудно сфокусировать на очень малом участке поверхности образца, обеспечив при этом достаточно высокую интенсивность. В промышленных приборах ЭСХА диаметр анализируемой области лишь совсем недавно был снижен до 1 мм, и потому этот метод является локальным лишь по глубине, а не по площади облучаемого объекта.
Чувствительность метода электронной спектроскопии значительно повышается при переходе к возбуждению оже-спектров электронным ударом: электронная оже-спектроскопил (ЭОС). Ожеспектрометры с электронным возбуащением в настоящее время выделились
в самостоятельную группу приборов, отличающихся от классических ЭСХА-спектрометров, кроме способа возбуждения электронных спектров, также более высоким вакуумом (давление остаточных газов в камере образца должно быть не выше 10-9 - 10-10 мм рт. ст. ).
Только в методе ЭОС с его высокой чувствительностью и большим отношением сигнала к шуму можно получать спектр за 0, 1 с и фиксировать его на экране осциллографа, что весьма существенно для изучения быстрых процессов на поверхности. Для ЭОС и ЭСХА эдектронная бомбардировка или облучение рентгеновскими квантами служат целям диагностики. Очистка же поверхности и послойное стравливание осуществляются при помощи специального источника, создающего пучок ионов или плазму. Такое разделение функций дает то важное практическое преимущество, что распыление образца можно в любое время прекратить для изучения особенностей состава поверхности па данном этапе.
Важным преимущество оже-спектроскопии (ЭОС) является возиожность использования хорошо сфокусированного луча, в результате чего можно исследовать отдельные мелкие частицы или анализировать поверхность образца от точки к точке. Основное достоинство метода ЭСХА в том, что получаемые спектры проще и их легче анализировать.
Изучение ультрадисперсных порошков никеля методом РФЭС объяснило их необычные магнитные свойства наличием тонкого слоя окисла, который невозмо; кно было обнаружить никакими другими методами [64]. Было также показано, чrо окисленный слой исчезает после отжига порошка при невысоких температурах в вакууме. Аналогично было обнаружено, что порошок нитрида титана с размером частиц 300 А имеет на поверхности оксинитридпую пленку толщиной 10 А, которая остается после высоко температурного вакуумного отжига [65].
ЭОС применяют как для качественного, так и для количественного анализа. Элементы, присутствующие на поверхности, идентифицируются по их электронным спектрам. Линии элементов, стоящих рядом в периодической системе Менделеева, хорошо разделены. Количественный анализ проводится по интенсивности пиков ивозможен при учете определенных поправок или когда имеются хорошие эталонные образцы. Наличие«химических» сдвигов для пиков одного и того же элемента позволяет определить его валентность и установить фазовый состав соединения, которое он образует.
Метод вторичнионной асс-спектрометрии (ВИМС) (4) или ионный микроанализ [48] основаны на принципе высокочувствительных масс-спектрометрических измерений и образований пучка ионов исследуемого вещества путем взаимодействия его с быстрыми ионами, энергии которых лежат в интервале от 10 до 20 кэВ. В качестве облучающих, или первичных, ионов используются ионы как инертных, так и химически активных газов. Для большинства аналитических работ выбирают ионы кислорода, так как это дает более интенсивный пучок вторичных ионов.
Правда, при этом становится невозможным обнаружение исходного кислорода в материале мишени.
Установка ВИМС состоит из четырех основных блоков: источника первичных ионов и системы формирования пучка, держателя образца и линзы, вытягивающей вторичные ионы, масс-спектрометра для анализа вторичных частиц по отношению массы к заряду (т/е) и высокочувствительной системы регистрации ионов. Для получения первичных ионов в большинстве установок используются газоразрядные или плазменные источники. Как и во всех методах микроанализа, установка содержит световой микроскоп для локализации образца.
Участок поверхности, с которого отбираются распыленные ионы для масс-анализа, в обычных промышленных приборах типа ионного зонда. может быть уменьшен до 1 мкм в диаметре. Поэтому создается возможность проводить локальный анализ и анализировать изолированные частицы или включения в них. Благодаря чрезвычайно высокой чувствительности этого метода (до 10-19 г) удается получать информацию о частице, меньшей сечения пучка.
Исследуемую частицу помещают на очень чистую металлическую подлоку, желательно содержащую только один изотоп и слабо ионизирующуюся при бомбардировке ионами. При работе с кислородными ионами лучше всего подходит сверхчистое золото и сверхчистый титан.
Метод ВИМС позволяет проводить качественный и количественный анализ, причем время проведения полуколичественного анализа составляет около одной минуты. Невысокая точность количестенного анализа объясняется трудностью создания надежных эталонов сложного состава для метода микроанализа. Приложения ВИМС можно разбить на пять широких (частично перекрывающихся областей): исследование поверхности, глубинные профили концентрации, распределение на поверхности, микроанализ и анализ объема твердого тела.
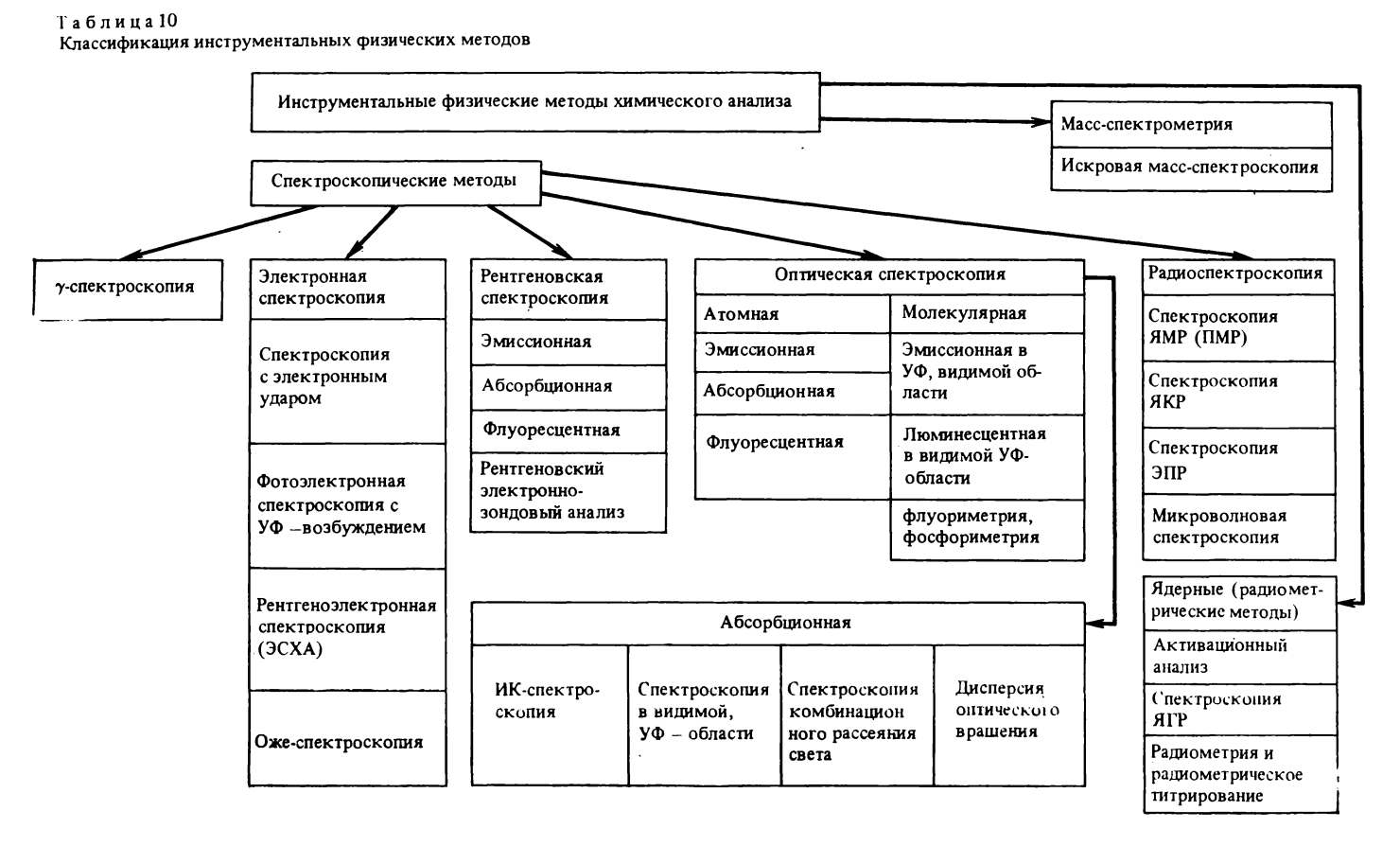
Классификация физических ипструмепталъных методов. В рамках книги невозможно подробно рассмотреть все известные в настоящее время методы инструментального химического анализа, а тем более проанализировать все огромное разнообразие типов, серий и модедей аналитических приборов, выпускаемых в Советском Союзе и зарубежными фирмами [40]. Каждому инструментальному методу химического анализа посвящены десятки отечественных и иностранных монографий [38, 40, 42-45, 51, 59, 60, 66]. Для многих инструментальных методов анализа выпускаются специализированны о научные журналы или имеются соответствующие разделы в журналах аналитического профиля. Здесь приводится классификация лишь основных инструментальных методов химического анализа (табл. 10) [40].
Роль физических методов анализа в современном химическом анализе очень значительна, а тенденция к их развитию растет с каждым годом.
Многочисленные физические методы анализа условно можно
разделить на три группы: спектрометрические, ядерно-физические (радиометрические) и метод масс-спектрометрии.
Инструментальные методы спектроскопии охватывают широкую область спектра электромагнитного излучения: от гамма-квантов и рентгеновских лучей до радиоволн сантиметрового и метрового диапазона, т. е. излучение с длиной волны от 10-2 до 109 нм (с частотой от 1019 до 108 Гц). В зависимости от типа энергетических переходов, применяемых источников излучения, способов разложения спектра и его регистрации спектроскопические методы принято условно разделять на следующие группы: гамма-спектроскопию, электронную, рентгеновскую, оптическую и радиоволновую спектроскопию.
Инструментальные методы спектроскопии позволяют получать информацию о химических свойствах различных веществ и материалов путем исследования процессов взаимодействия электромагнитного излучения с атомами и регистрации этих взаимодействий в виде электрических сигналов, частота и интенсивность которых непосредственно связаны с качественными и количественными характеристиками химического состава и строения исследуемого образца.
Как правило, спектроскопию у-квантов включают в ядернофизические методы аналитической химии, рассматривая ее как один из методов радиометрического анализа, основанный на использовании взаимодействия ядерного у-излучения с электронами атомных и молекулярных оболочек, и называют гамма-спектроскопией [38].
Электронная спектроскопия, включающая четыре основных метода: спектроскопию электронного удара, фотоэлектронную спектроскопию в вакуумной ультрафиолетовой области, рентгеноэлектронную спектроскопию (электронная спектроскопия для химического анализа) и оже-спектроскопию, - основана на изменении кинетической энергии электронов, что позволяет определять потенциалы ионизации атомов и молекул, а тем самым получать. величины энергии связей электронов как внутри отдельных атомов, так и внутри атомов, входящих в состав молекулы [5, 59, 60].
Масс-спектрометрию часто объединяют со спектроскопическими методами, основываясь на процессах взаимодеиствия ионизирующих частиц и электромагнитных полей с атомами имолекулами и на формальном сходстве образующихся массспектров с эмиссионными спектрами: сигналы, составляющие массспектр, в соответствии с величинами т/е (т - масса иона, е - его заряд) характеризуются их местоположением в спектре (частотой) и интенсивностью (количеством) образующихся ионов. Однако аналогия масс-спектроскопии со спектроскопией все же не правомерна. В отличие от спектроскопии разнообразные методы массспектрометрии вызывают не только возбуждение и ионизацию исследуемых молекул, но и, что является основой метода массспектрометрии, расщепление молекулярных ионов на фрагменты -
осколочные ионы. Следовательно, совокупность процессов при масс-спектроскопическом исследовании веществ приводит к необратимому изменению исходного состояния молекул; по своей природе метод масс-спектрометрии является разрушающим.
Ближе всего связь масс-спектрометрии, с одной стороны, с электронной спектроскопией, а с другой - с ионизационными методами, так как все эти методы объединяют процессы, проте- кающие с участием материальных частиц (квантов, электронов, ионов).
Широко используемые в настоящее время в аналитической химии ядерные (радиометрические) методы элементного анализа основаны на измерении наведенной (радиоактивационный анализ) и естественной (методы радиоактивных индикаторов, или меченых атомов) радиоактивности.
В активационном анализе образец облучают потоком нейтронов заряженных частиц (протонов, ионов 3Не) и у-лучами для образования радиоактивных изотопов (из стабильных). Измерение величины наведенной радиоактивности позволяет определить количество исследуемого элемента в образце.
Радиометрические методы химического анализа хотя и не от носятся к инструментальным, но тем не менее определение следовых количеств элементов с их помощью базируется на измерении величины радиоактивности.
В число ядерных инструментальных методов химического анализа следует включить и ядерную гамма-резонансную спектроскопию (мессбауэровскую спектроскопию).
Методы исследования элементного состава одни и те же как для монолитов, так и для порошков, однако имеется ряд методических особенностей, которые необходимо учитывать при исследовании порошков. Обнаружено, например, что на результаты оптического спектрального анализа легированных порошков влияет их дисперсность [65]; результаты количественного рентгеноспектрального анализа смесей металлических порошков могут быть искажены, если исследуемые порошки имеют различную пластичность. Влияние пластичности проявляется при изготовлении объекта для исследования в виде таблетки путем прессования. В поверхностном слое состав смеси обогащается в сторону более пластичной компоненты [69].
По ИК-спектрам была сделана оценка чистоты проб и прочности межатомных связей ультрадисперсных порошков нитрида алюминия, полученных различными способами [70].
Изучение ультрадисперсного порошка олова методом ЯГР
(эффект Мессбауэра) обнаруживает в нем наличие окислов
Sn02 и SnO (в количественном соотношении 2: 1) [27].
Электронный парамагнитный резонанс широко применяется для изучения неметаллических порошков. Он позволяет изучать состояние поверхности, возникающей в процессе диспергирования. наблюдать разорванные связи, исследовать дефектную структуру кристаллических частиц [71].
Определение содержания газов в объеме и па поверхности частиц порошков. В то время как физические свойства компактных материалов зависит в основном от содержания газов в объеме вещества, для порошков доминирующее значение могут иметь газы, сорбированные на поверхности. Поэтому для диагностики порошков, применяемых в порошковой металлургии, очень важно раздельное определение газов в объеме ина поверхности частиц порошка.
В отличие от компактных металлов металлические порошки как объект анализа имеют ряд особенностей: 1) порошки разных фракций одной и той же партии, как правило, отличаются по содержанию газов, поэтому для получения достоверных данных о среднем содержании газов в партии необходимо провести большое число измерений; 2) общее объемное и поверхностное содержание газов в порошках велико (обычно более 1- 10-2 мас %); 3) некоторые порошки обладают сильно развитой поверхностью, сорбирующей газы из окружающей среды в процессе изучения и хранения порошков. Количестно газов на поверхности частиц таких порошков может быть больше, чем в объеме [72].
Для определения содержания водорода, азота, кислорода, а также углерода в металлических порошках используется большое число аналитических методов. Кроме различий между поверхностной и объемной концентрациями, при анализе необходимо учитывать, в каких формах существуют газы в металлическом порошке: растворяются ли они в металлической решетке, или присутствуют в качестве соединений (оксидов и т. п. ). В большинстве аналитических методов газ определяется в той или иной форме.
Важнейшие методы и границы их точности приведены в табл. 11 [73].
Выбор одного из аналитических методов зависит от анализируемого металла и от предъявляемых требований. Метод горячей экстракции выгодно отличается тем, что водород, азот и кислород могут быть определены одновременно или последовательно один за другим. Метод горячей экстракции иметоды второйгруппы используются для абсолютного определения содержания газов и углерода в порошках. На практике эти аналитические методы наиболее распространены для научных исследований.
К третьей группе относятся прямые физические методы. Эти методы весьма сложные и в настоящее время используются только в спорных случаях для особо ответственного анализа.
Метод горячей экстракции или восстановительного плавления заключается в выделении содержащегося в пробе водорода, азота и кислорода в вакуум или газовую среду при высоких температурах в пристутствии углерода. Образцы можно исследовать как в твердом, так и в расплавленном состоянии; наиболее часто образцы переводятся в расплав, так как при этом существенно ускоряется процесс дегазации. Выделенные газы собираются, разделя ются и количество их определяется методами газового анализа. Аппаратура метода содержит два основных узла: узел выделения
|
газа из образца (•печь), где образец нагревается до температуры, необходимой для выделения определенного газа, и анализатор, предназначенный для анализа выделенных газов [74, 75].
Для анализа металлических порошков с использованием горячей экстракции образцы удобно взвешивать и нагревать в одном и том же закрытом графитовом контейнере. Предварительное прессование, заворачивание в металлическую фольгу ипримене ние металлических капсул снижают точность анализа [72].
Анализ выделенного газа можно проводить хроматографически с помощью разделительной колонки и детектора, а для малых количеств газа масс - спектрометрически с замером парциальных давлений газа. Применяют также кулонометрию, кондуктометрию, эмиссионную спектроскопию.
Методы горячей экстракции используют наиболее часто для определения газов в металлических порошках, так как они универсальны идают результаты за короткий промежуток времени. Метод горячей экстракции пригоден для определения общего содержания газа в образце. Анализ образцов одного и того же материала с различным отношением площади поверхности к объему позволяет оценить объемное и поверхностное содержание газов расчетным путем [72].
Достаточно прост и удобен для применения к порошкам метод восстановления водородом для определения содержания кислорода и водяного пара, адсорбированных на поверхности.
При химических методах анализа всегда определяется только один элемент, поэтому их используют в тех случаях, когда применение способа горячей экстракции затруднено (например, при анализе азота), а химический анализ (например, на углерод) является наиболее простым. Существуют индивидуальные методикидля определения кислорода, азота иуглерода, имеющие широкое распространение.
Физические методы позволяют определять газы иуглерод непосредственно в пробе, но дают только относительные данные, так как при всех анализах необходимо устанавливать калибровочные кривые для зависимости между измеренными значениями и содержанием газа в стандартной пробе.
Нейтронный активационный анализ используется для определения кислорода, азота и углерода. Применяются все три известных активационных метода с быстрыми нейтронами, высоко-энергетическими у-квантами и заряженными частицами. Быстрые нейтроны и у-кванты глубоко проникают в образец и дают возможность определять содержание газа в объеме, в то время как анализ с помощью заряженных частиц позволяет определять газы и углерод толыю в приповерхностной зоне и на поверхности [76]. Метод активационного анализа в сочетании с горячей экстракцией позволяет определять содержание кислорода и азота в
объеме с точностью до ~10-6 мас. %.
В полученных плазменным распылением порошках W и Мо изучалось содержание кислорода методом нейтронного активационного анализа, который позволил связать высокое содержание кислорода с большой адсорбционной способностью плазменных порошков, проявляющейся сразу после их получения [77].
Глава седьмая
|
|
|
