 |


платиновыми электродами (кондуктометрическая ячейка)
|
|
|
|
на разных светофильтрах
| Длина волны светофильтра, нм | ||||||
| Абсорбция, отн.ед. |
Для работы выбран светофильтр с l = … нм.
3) Построение калибровочного графика
Таблица 2 – Результаты фотометрирования стандартных
и анализируемого раствора
| № колбы | Концентрация раствора, моль/дм3 | Абсорбция раствора, отн. ед. аб-ции. |
| Анализир. раствор |
По полученным результатам строят калибровочный график на миллиметровой бумаге и определяют концентрацию железа в анализируемом растворе.
|

Результаты проверяют у преподавателя или лаборанта, оформляют отчет по лабораторной работе.
5 ПЛАМЕННАЯ ФОТОМЕТРИЯ
5.1 Теоретические основы
Метод пламенной фотометрии является одним их видов эмиссионной спектроскопии. Он основан на измерении интенсивности излучения, испускаемом анализируемыми элементами в пламени. Чувствительность метода определяется температурой пламени и условиями возбуждения атомов. В качестве источников возбуждения атомов используется пламя различных типов. Средняя температура некоторых наиболее широко распространенных видов источников возбуждения приведена в таблице 5.1.
Таблица 5.1 - Основные источники возбуждения атомов
| Источник возбуждения | Горючий газ | Окислитель | Температура, К |
| Пламя | Пропан-бутан | Воздух | |
| Ацетилен | Воздух | ||
| Ацетилен | Кислород | ||
| Ацетилен | Закись азота | ||
| Вольтовая дуга | - | - | |
| Конденсированная искра | - | - | |
| Лазерное излучение | - | - |
Вследствие невысокой температуры пламени горючих смесей, их эмиссионные спектры состоят их небольшого числа спектральных линий и имеют низкие потенциала возбуждения. Это позволяет использовать при анализе фотометры, снабженные интерференционными светофильтрами.
|
|
|
Атомные спектральные линии в пламени излучают щелочные и щелочноземельные металлы, галлий, кадмий, хром, марганец, никель, кобальт, медь, серебро и другие элементы. В настоящее время методом пламенной фотометрии можно определять до 40 элементов.
Набор частот, соответствующий спектру каждого элемента, определяется структурой внешней оболочки атома и, следовательно, положением элемента в таблице Менделеева. Вследствие этого не может быть случаев полного совпадения эмиссионных спектров различных веществ.
Качественный анализ анализируемых веществ основан на наличии в спектре излучения характеристических линий, присущих данному элементу. В пламенной фотометрии качественный анализ проводится с использованием набора светофильтров.
В основе количественного анализа лежит прямо пропорциональная зависимость интенсивности спектральных линий от числа возбужденных частиц, и, следовательно, от концентрации определяемого элемента при постоянных температуре и условиях атомизации. Эта зависимость выражается уравнением Ломакина:
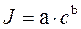 , (5.1)
, (5.1)
где I – интенсивность спектральной линии; с – концентрация элемента в пробе; a – коэффициент, зависящий от режима работы источника возбуждения; b – коэффициент самопоглощения.
Определяемые элементы поступают в пламя в виде аэрозоля, полученного при распылении анализируемых растворов воздухом в распылительной камере.
В пламени протекает ряд следующих процессов:
1) Растворитель испаряется или сгорает, а в пламени остаются мельчайшие частицы твердого образца;
2) Твердые вещества переходят в газообразное состояние (превращаются в пар), то есть образуется атомный пар;
3) Возбуждение атомов под действием энергии пламени. Возбуждение газообразных атомов связано с переходом одного из электронов в более высокое энергетическое состояние;
|
|
|
В условиях термодинамического равновесия доля возбужденных атомов определяется уравнением Больцмана:
 , (5.2)
, (5.2)
где N * – количество возбужденных атомов в пламени; N 0 – общее количество атомов в пламени; Δ E – потенциал возбуждения, Дж; k – постоянная Больцмана (k =1,380662 10-22 Дж К); T – абсолютная температура, К.
Согласно уравнению Больцмана, только небольшая часть газообразных атомов переходит в возбужденное состояние и доля возбужденных атомов быстро снижается при возрастании потенциала возбуждения элементов. Например, при одинаковой температуре доля возбужденных атомов натрия в 107 раз превышает долю возбужденных атомов цинка. Вследствие этого для определения цинка требуется более высокая температура. При повышении температуры число возбужденных атомов увеличивается.
4) Излучение света возбужденными атомами: при возвращении электрона из возбужденного состояния на более низкий энергетический уровень или в основное состояние испускается ультрафиолетовое или видимое излучение. Интенсивность и частота излучения зависят от природы, концентрации анализируемого элемента в растворе. Частота испускаемого излучения зависит от разности энергий двух состояний атома:
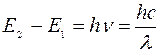 , (5.3)
, (5.3)
где E 2 – энергия более высокого возбужденного состояния, Дж; Е 1 – энергия более низкого возбужденного или основного состояния, Дж; h – постоянная Планка; ν – частота испускаемого ультрафиолетового или видимого света, Гц; с – скорость света, м/с; λ – длина волны излучаемого света, м.
5.2. Схема пламенного фотометра
Схема пламенного фотометра представлена на рисунке 5.1.
|
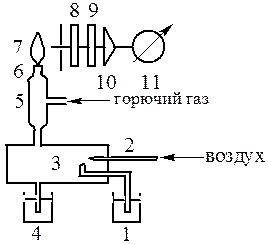
Рисунок 5.1 - Схема пламенного фотометра ПФЛ-1:
1 – стаканчик с раствором; 2- распылитель; 3 – распылительная камера; 4 – водяной затвор; 5 – смесительная камера; 6 – горелка; 7 – пламя; 8 – входная щель; 9 – светофильтр; 10 – фотоэлемент; 11 – усилитель;
12 – гальванометр
В качестве монохроматора в приборе использованы три сменных светофильтра, полосы пропускания которых соответствуют аналитическим спектральным линиям натрия, калия и кальция [1, табл. 8]. Полосы пропускания светофильтров являются достаточно узкими. Светофильтры пропускают излучение аналитической линии определяемого элемента и поглощают излучение других элементов.
|
|
|
В качестве аналитического сигнала при количественном анализе в пламенной фотометрии используются показания гальванометра 12, расположенного на передней панели прибора. Гальванометр измеряет ток фотоэлемента 10, который зависит от интенсивности спектральной линии и, в свою очередь, от концентрации анализируемого раствора.
5.3 Порядок работы на пламенном фотометре
1) Фотометр включает лаборант за 10 минут до начала работы.
2) Устанавливают светофильтр, соответствующий анализируемому элементу, поворачивая рычаг на правой стенке прибора.
3) Проводят калибровку прибора. Для выполнения калибровки необходимо подготовить стандартные растворы.
Лабораторная работа № 15
ОПРЕДЕЛЕНИЕ ЭЛЕМЕНТОВ
МЕТОДОМ ПЛАМЕННОЙ ФОТОМЕТРИИ
| Цель работы: | определение концентрации ионов Na+, K+, Ca2+ в анализируемой смеси. |
| Реактивы: | стандартные растворы хлоридов натрия, калия, кальция. |
| Посуда: | мерные колбы вместимостью 50 см3 (13 шт.), капельница, пробки. |
Выполнение работы:
1) Готовят серии стандартных растворов солей каждого определяемого элемента с различными фиксированными концентрациями, внося в мерные колбы на 50 см3 заданный лаборантом объем исходного раствора. Объем растворов доводят дистиллированной водой до метки. Растворы в колбах тщательно перемешивают.
2) Пробу для анализа студенты получают у лаборанта в мерной колбе и также доводят до метки дистиллированной водой.
3) Заполняют нумерованные стаканчики приготовленными стандартными, анализируемыми растворами и дистиллированной водой.
4) Устанавливают светофильтр, соответствующий анализируемому элементу, поворачивая рычаг на правой стенке прибора.
5) Трубку распылителя опускают в стаканчик с дистиллированной водой и с помощью ручки гальванометра, расположенной на передней панели прибора слева, устанавливают стрелку гальванометра на ноль.
|
|
|
6) Трубку распылителя опускают в стаканчик с самым концентрированным раствором, при этом стрелка прибора отклоняется вправо. С помощью правой ручки гальванометра устанавливают стрелку в положение, соответствующее делению шкалы 80-90. Повторяют процедуру 3-4 раза. Такая методика гарантирует от зашкаливания прибора при дальнейших измерениях, т.к. все растворы с меньшей концентрацией соответствуют показаниям прибора между нулем и максимальным показанием гальванометра.
7) Снимают показания гальванометра для остальных растворов анализируемого элемента в порядке снижения концентрации. Затем снимают показания гальванометра анализируемой смеси. Результаты заносят в таблицу 1.
8) По полученным экспериментальным данным строят калибровочный график. Для этого по оси абсцисс откладывают значения концентрации растворов в единицах мг/мл, по оси ординат – соответствующие значения показаний гальванометра.
9) По калибровочному графику определяют концентрацию ионов анализируемого металла.
10) Повторяют измерения с п.3 по п.7 для других солей.
Для непрерывного контроля результатов рекомендуется наносить значения концентрации и показания прибора на калибровочный график одновременно с записью в таблицу. В этом случае можно проверить точки, выпавшие из графической зависимости, вновь установив стаканчик с раствором для измерения.
Расчет результатов анализа
1) Расчет концентрации приготовленных стандартных растворов солей
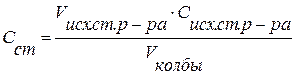 ,
,
где Сисх.ст. р-ра – концентрация исходного стандартного раствора соли в склянке, мг/мл; Сст. – концентрация, приготовленного раствора в колбе, мг/мл; Vисх.ст.р-ра – объем исходного стандартного раствора, отмеренный по бюретке, мл; Vк – объем колбы, мл.
2) Экспериментальные результаты заносят в таблицу 1.
Таблица 1 – Результаты эксперимента
| № колбы | Показания гальванометра | |||||
| Na | K | Ca | ||||
| C, мг/мл | J, мА | C, мг/мл | J, мА | C, мг/мл | J, мА | |
| Анализир. раствор |
Построение калибровочного графика и определение концентрации металла в анализируемом растворе.
Калибровочный график строится по результатам, приведенным в таблице 1.
|
|
|
|
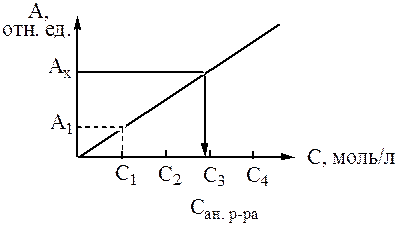
Результат проверяют у преподавателя или лаборанта, оформляют отчет по выполненной работе.
2 ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
2.1 Кондуктометрический метод анализа
2.1.1 Теоретические основы
Электропроводность раствора электролита G и его концентрация С связаны между собой функциональной зависимостью. При низких концентрациях
|
|
|
 , (2.1)
, (2.1)
где k – некоторая постоянная величина.
Для правильного построения методики кондуктометрического анализа необходимо знать электрические свойства растворов, термины, определения, физико-химические основы переноса зарядов в электролитах, причины нелинейности электропроводности [3].
Электропроводность растворов в системе СИ G измеряется в сименсах (См). Различают удельную χ  и эквивалентную λ электропроводность раствора. Удельная электропроводность См/см или Ом-1·см-1 – электропроводность 1 см3 раствора, находящегося между электродами площадью 1 см2 каждый, расстояние между которыми равно 1 см. Эквивалентная электропроводность – электропроводность слоя электролита, содержащего 1 эквивалент вещества, измеренная при расстоянии между электродами 1 см. Удельная и эквивалентная электропроводность связаны соотношением
и эквивалентную λ электропроводность раствора. Удельная электропроводность См/см или Ом-1·см-1 – электропроводность 1 см3 раствора, находящегося между электродами площадью 1 см2 каждый, расстояние между которыми равно 1 см. Эквивалентная электропроводность – электропроводность слоя электролита, содержащего 1 эквивалент вещества, измеренная при расстоянии между электродами 1 см. Удельная и эквивалентная электропроводность связаны соотношением
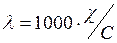 ,
,
где С – молярная концентрация эквивалента, моль/дм3 .
Эквивалентная электропроводность – аддитивная величина, которая выражается соотношением
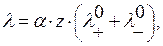
где α – степень диссоциации;
z – заряд иона;
 и
и 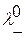 - подвижности положительных и отрицательных ионов.
- подвижности положительных и отрицательных ионов.
В свою очередь, 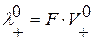 ;
; 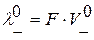 , где F – число Фарадея,
, где F – число Фарадея, 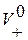 и
и 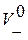 – абсолютные скорости движения ионов (в см/с) в поле напряженностью 1 В/см. В таблице 2.1 приведены подвижности ионов в водных растворах при бесконечном разбавлении при t =25˚С.
– абсолютные скорости движения ионов (в см/с) в поле напряженностью 1 В/см. В таблице 2.1 приведены подвижности ионов в водных растворах при бесконечном разбавлении при t =25˚С.
Таблица 2.1 - Подвижности ионов в водных растворах
при бесконечном разбавлении при t=25˚С
| Катионы |
| Анионы |
|
| H+ | 349,9 | OH- | 198,4 |
| K+ | 73,5 | Cl- | 76,4 |
| Na+ | 50,1 | Br- | 78,2 |
| Li+ | 38,7 | J- | 76,8 |
| ½ Ca2+ | 59,5 | NO3- | 71,5 |
| ½ Mn2+ | 53,8 | CH3COO- | 40,9 |
| ½ Zn2+ | 53,5 | H2PO4- | 36,0 |
| NH4+ | 73,5 | ½ SO42- | 80,0 |
Подвижности ионов определяют значение k в (2.1) и вид кривых титрования.
Измерение электропроводности растворов и величин, связанных с электропроводностью, лежит в основе кондуктометрического метода анализа. Метод реализуется двумя вариантами применения.
1. Прямая кондуктометрия, основанная на измерении электропроводности растворов по уравнению (2.1). Аналитическим сигналом является G, а k – коэффициент чувствительности.
В данном методе необходимо знать функциональную зависимость G от C. Ее устанавливают, измеряя электропроводность стандартных растворов с известными концентрациями, и выражают в виде калибровочной характеристики: в виде графика, таблицы или информации, введенной в ЭВМ.
2. Кондуктометрическое титрование, в котором по изменению электропроводности анализируемого раствора определяют точку эквивалентности.
В кондуктометрическом титровании вид функциональной зависимости не имеет существенного значения, важен перелом на кривой кондуктометрического титрования, соответствующий точке эквивалентности.
Измерение электропроводности растворов
(прямая кондуктометрия)
Благодаря простоте аппаратурного оформления, возможности определения электропроводности в мутных, окрашенных и вязких растворах кондуктометрия широко применяется в химических лабораториях и на производстве. С помощью кондуктометрии можно быстро и с малыми погрешностями простым измерением электропроводности найти концентрацию раствора. С помощью кондуктометрического метода определяют также растворимость, константу и степень диссоциации и другие важные физико-химические свойства электролитов. Типичная калибровочная кривая изображена на рисунке 2.1, стрелками показан способ нахождения концентрации. Точками отмечены измерения электропроводности стандартных растворов. Начальная часть графика представляет собой прямую линию, выходящую из начала координат (при учете собственной электропроводности растворителя).
|

Рисунок 2.1 – Калибровочная кривая метода прямой кондуктометрии
Кондуктометрическое титрование
В кондуктометрическом титровании могут использоваться реакции кислотно-основного взаимодействия, осаждения, комплексообразования. Преимуществом метода кондуктометрического титрования является возможность дифференцированного определения веществ в многокомпонентных системах и в окрашенных и мутных растворах. Кондуктометрическое титрование индивидуальных электролитов может быть проведено с относительной ошибкой ± 1 %, при титровании смеси электролитов – ± 2%, нижний предел определяемых концентраций – 10-4 моль/дм3.
Кондуктометрическое титрование протолитов и их смесей
Электропроводность растворов сильных кислот и сильных оснований определяется в основном ионами H3O+ (в кислоте) или OH- (в сильном основании), т.к. они обладают наибольшей подвижностью (см. таблицу 2.1). По мере прибавления титранта электропроводность, замеряемая после добавления каждой порции титранта, понижается вследствие замены высокоподвижных ионов менее подвижными катионами основания (или анионами кислоты). После достижения точки эквивалентности электропроводность повышается вследствие накопления ионов OH- или H3O+ титранта. Точка эквивалентности определяется графически экстраполяцией прямых до их пересечения (рисунок 2.2, а).

Рисунок 2.2 – Кривые кондуктометрического титрования:
а – сильная кислота титруется сильным основанием 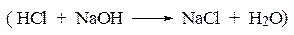
б – слабая кислота титруется сильным основанием

При титровании слабых протолитов электропроводность вначале имеет значительно меньшую величину, обусловленную низкой степенью диссоциации слабого электролита. Вблизи точки эквивалентности наблюдается более размытый переход от одного участка кривой к другому (рисунок 2.2, б).
Возможность анализа смеси протолитов зависит от концентрации протолитов и соотношения констант диссоциации (рисунок 2.3). При титровании смеси сильного и слабого протолитов наблюдаются 2 точки эквивалентности, если соблюдается неравенство: К 1: К 2 > 104, где К 1 и К 2 - константы диссоциации сильного и слабого протолитов.

Рисунок 2.3 - Кривая кондуктометрического титрования смеси кислот
В этом случае первым титруется сильный протолит, электропроводность значительно понижается, затем титруется слабый электролит, электропроводность незначительно повышается за счет накопления ионов с невысокой подвижностью (катионов и анионов). После достижения второй точки эквивалентности электропроводность раствора увеличивается вследствие возрастания концентрации ионов OH- или (H3O+) с высокой подвижностью.
2.1.2. Аппаратура кондуктометрических измерений
Для определения электропроводности растворов электролитов преимущественное распространение получил мостовой метод, цепь моста питается переменным током. Измерение проводится по обычной схеме моста Уитстона (рисунок 2.4), в котором rx - ячейка, состоящая из стеклянного сосуда и двух плоскопараллельных жестко закрепленных электродов (рисунок 2.5), r 2, r 3, r 4 – постоянные сопротивления; Г – измерительный прибор, отградуированный в единицах электропроводности, См. При кондуктометрическом титровании используется микробюретка емкостью 10 см3 , которая устанавливается над ячейкой.
Кондуктометр марки ОК-102/1 включают в электрическую сеть. Тумблер включения ставят в положение “вкл.”. Прибор прогревается в течение 15 мин. Выключают прибор только после окончания работы.

Рисунок 2.4 – Схема моста Уитстона
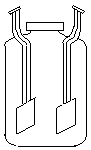
Рисунок 2.5 – Схема сосуда с прочно закрепленными
платиновыми электродами (кондуктометрическая ячейка)
2.1.3 Методика кондуктометрических измерений
Бюретку заполняют стандартным раствором титранта. В электролитическую ячейку переносят аликвотную часть анализируемого раствора, доводят его объем дистиллированной водой на 0,5 см выше электродов. Ставят ячейку на магнитную мешалку и присоединяют к прибору. Устанавливают стрелку прибора на крайнее максимальное деление шкалы, записывают первый отсчет. Последовательно приливая из бюретки по 0,2 см3 титранта, записывают показания прибора в таблицу 2.2.
Таблица 2.2 – Экспериментальные данные
| Объем прибавленного титранта, мл | G, См |
Затем строят кривую титрования, откладывая на оси ординат электропроводность (см. рисунок 2.2). На оси абсцисс – объем титранта. Точка эквивалентности определяется графически на пересечении прямых. Зная объем и концентрацию титранта, рассчитывают содержание анализируемого вещества.
2.3 Потенциометрический метод анализа
2.3.1 Теоретические основы
Потенциометрические методы анализа основаны на использовании зависимости ЭДС электрохимической ячейки от активности определяемого иона в растворе.
Простейшая электрохимическая ячейка содержит два электрода: индикаторный, потенциал которого прямо или косвенно зависит от концентрации потенциалопределяемого иона, и электрода сравнения.
ЭДС = Е 1 – Е 2, (2.2),
где ЭДС – электродвижущая сила,
Е 1 и Е 2 – потенциалы электродов, индикаторного и сравнения соответственно.
Математическое выражение зависимости электродного потенциала от активной концентрации ионов, относительно которых обратим электрод, даётся уравнением Нернста:
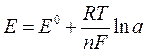 ,
,
где Е0 – стандартный потенциал системы;
R – газовая постоянная (8,312 Дж/моль·К);
T – абсолютная температура;
F – постоянная Фарадея (96500 Кл/моль);
п – число электронов, участвующих в электродной реакции;
a – активная концентрация определяемого иона.
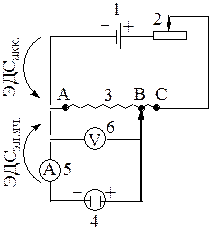
Рисунок 2.8 - Принципиальная схема измерения потенциала:
1 – источник с известной ЭДС (аккумулятор); 2 – постоянное сопротивление;
3 – реохорд 4 – электролитическая ячейка; 5 – амперметр; 6 – вольтметр
Потенциометрический метод анализа подразделяется:
1) на прямую потенциометрию (ионометрия);
2) потенциометрическое титрование в водных и неводных средах.
Прямая потенциометрия с применением
|
|
|
