 |


ЭКСП метод опред порога коагул
|
|
|
|
Диссолюционная пептизация
Свежий осадок, содержащий ДЭС, просто промывают большим кол-вом воды. При этом проис размывание АСПИ и часть ионов этого слоя переходит в Ср, образуя ДСПИ. Коллоидная ч-ца при этом приобретает заряд и образ-ся в рез-те дис-ой пептизации из свежего осадка золь приобр относит уст-сть: {[Fe(OH)3] m n Fe3+ 3 n Cl–}0 + H2O → {[Fe(OH)3] m n Fe3+ (3 n – x)Cl–} x + x Cl–,
{[Fe(OH)3] m n OH– n K+}0 + H2O → {[Fe(OH)3] m n OH– (n – x)K+} x – x K+.
В данном случае заряд кол-ых ч-ц определяется не приро-
дой пептизатора (в кач к-ого в обоих рассм-ых случаях
выступает обычная вода), а зарядом ПОИ, входивших в состав исходных частиц гидрозоля.
При химической пептизации к ч-цам свежеполученного свежеобразовавшегося) осадка, не содержащего ДЭС, добавляют эл-т, ионы к-ого сами не способны адсорбир-ся на незаряж-ой пов-сти ч-ц осадка; но в рез-те хим взаимод электролита-пептизатора с ч-цами осадка происх раств-ие их тонкого пов-ого слоя, в рез-те чего в среде обр-ся ПОИ, способн стабилизировать ч-цы гидрозоля:
{[Fe(OH)3] m }0 + 3 n HCl → {[Fe(OH)3] m–n }0 + n FeCl3 + 3H2O,
{[Fe(OH)3] m–n }0 + n FeCl3 → {[Fe(OH)3] m–n n Fe3+ (3 n – x)Cl–} x + x Cl–.
Путем диссол пептизации во взвешенное состояние м.б.
переведены свежие осадки, образов-еся в рез-те конц-нной коагуляции (т.к. только в этом случае ч-цы сохраняют ДЭС).Если осадок образ-ся в рез-те нейтрализац коагуляции, то для его перевода во взвешенное состояние необх использовать адсорбц-ую или хим-ую пептизацию.
41. Виды устойчивости дисперсных систем
Уст ДС – постоянство их св-в во времни.Виды уст-ти по отн к укрупнению и к осаждению.сущ-ет:седиментационная(СУ) и агрегативная (АУ). СУ – спос-сть ДС сохр равномерное и пост во времени распределение ч-ц ДФ по всему объему ДСр, т.о. СУ – это уст-сть ДС к разделению фаз. Потеря седим-ой уст-сти приводит к оседанию (ρДФ > ρДСр, прямая седиментация) или всплыванию ч-ц ДФ (ρДФ < ρДСр, обратная седиментация); индив-сть ч-ц ДФ при седиментации в общем случае сохр. АУ - способность ДС сохранять пост во времени дисп-сть и индив-сть ч-ц ДФ.
|
|
|
От усл обр ДС: лиофильные (интенсивн взаимод ч-ц ДФ и ДСр,обр при самопроизв диспергир одной из фаз, явл ТД и агрегативн уст) и лиофобные (агрег неуст,избыт пов-ной эн, самопроизв укрупн ч-ц ДФ, сниж пов эн за счет уменш площади межфазн пов-ти). Укрупн ч-ц 2споб: 1. изотермич перегонка -перенос в-в от мелких ч-ц к крупн(мелк испар, а крупн-растут) При повыш Т- рекристализаия. Движущ силой обоих – разл хим потенциал мелких и крупных. Коагуляция – отдельные ч-цы ДФ слипаются(не теряют своей индивид, м-у ч-ками сущ тонкие послойки). Коагул иногда приводит к расслоению фаз. Гомо- слипание ч-ц ДФ, имеющ одинак природу(одинак заряд), гетеро- различн природу(противопол заряж) Адагуляция – адгезионное взаимод ч-ц ДФ с макропов-тью (т.е.ч-цы ДФ слипаются не друг с др, а с пов-тью раздела). Коалесценц –(полное) слияния ч-ц ДФ. Структурообр – в конц ДС потеря сист агрегативн уст-ти (ч-цы ДФ, раздел послойками). Флокуляция – потеря уст-сти под действием высокомолек соедин(ВМС).При флок теряется агрегатив,затем седимент уст-ть.образ рыхлые хлопьевидн стр-ры – флокулы(неорг полимеры,орган высокомол соедин)синтетич(неионоген и полиэлектролиты(анион и катионные)). Протек по 2 мех-ам: 1нейтрализ-ый:осущ действ макроионов полимера,имеющ заряд, противопол по знаку заряду ч-цы:макроионы катион флоукл адсорбир на отриц заряж участках) 2.мостичный – вначале происх адсорб макромол-л на одной частице ДФ.мол флок закрепляется частью своих сегментов на пов-ти частиц. Не связ с данной частиц сегменты макромол-л закрепл на свободн пов-ти близлежащ частицобр мостики между частицами ДФ.
|
|
|
42 Факторы устойчивости дисперсных систем
ТД и кинетические факторы уст-сти. Уст-сть ДС (при сохранении площади межфазной пов-сти), обесп те, к-ые снижают пов-ное натяжение - ТД, они уменьшают вероятность эффективных столкн ч-ц ДФ(создают потенц барьеры, замедляющие или даже исключающие процесс коагуляции). Кинетические факторы снижают скорость коагул и связаны с гидродинамич св-вами ДСр: с замедл дв-ия ч-ц ДФ, вытекания и разрушения прослоек м-у ними.
К ТД факторам уст-сти относят электростатический, адсорбционно-сольватный и энтропийный факторы, к кинетическим– стр-рно-механич и гидродинамический.
Электростатический фактор - в ум-нии межф пов-ого натяжения за счет возникн ДЭС на границе раздела фаз. Чем больше заряды внутр и внешней частей ДЭС, тем бо-
лее агрегативно уст-вой явл-ся ДС.
Адсорбционно-сольватный фактор заключ в ум-нии межфазного натяжения при взаимод-ии ч-ц ДФ с ДСр (благодаря адсорбции и сольватации) в соотв-ии с ур-ем Дюпре для р-ты адгезии и адсорбц-ым ур-ем Гиббса. Если в ДС вводить ПАВ (низкомолекулярные), то их мол-лы (или ионы), адсорбируясь на пов-сти ч-ц ДФ, снижают пов-ое натяжение на межфазной границе и ув-ют агрег уст-сть ДС.
Энтропийный фактор действует в сист, в к-ых ч-цы
или их поверхностные слои участвуют в тепловом дв-ии. Сущность его состоит в стремлении ДФ к равномерному
распределению по всему объему ДС. Если на пов-сти ч-ц ДФ адсорбируются мол-лы(или ионы) среднецепочечных ПАВ или ВМС, то стремление УВ хвостов этих мол-л (ионов) к наиб свободе (беспорядку) также повышает уст-сть ДС за счет энтропийного фактора.
Действие структурно-механич фактора обусловлено тем,
что образующиеся вследствие адс-ции ионов, м-л ПАВ и ДСр на пов-сти ч-ц ДФ пленки обладают упругостью и механич прочностью; разрушение этих пленок требует затрат эн и времени, что замедл процесс агрегации ч-ц ДФ
Гидродинамический фактор снижает скорость коагуляции благод ув-ию вязкости ДСр и плотностей ДФ и
ДСр.Введение в ДС сахаров (явл-хся пов-но-неактивными, т.е. не способными адсорбироваться на пов-сти раздела фаз) и часто используется для ув-ия агрегативной уст-сти
ДС разл природы. Наиб высокая уст-сть ДС наблюдается при сов-сти действия ТД и кинетических факторов, когда наряду со снижением межфазного натяжения проявл стр-но-механические св-ва межчастичных прослоек.
|
|
|
метод нейтрализации уст: действие электростатического фактора снижается при введении в систему электролитов, вызывающих сжатие ДЭС. Сольватация при адсорбционно-сольватном факторе м.б. исключена лиофобизацией ч-ц ДФ при помощи соотв-щих в-в. Действие стр-но-механич фактора можно уменьшить при помощи в-в, разжижающих и растворяющих упр стр-рирован слои на пов-сти ч-ц ДФ. Разбавление гидрозоля водой будет приводить к размыванию адсорбц-ого слоя ПИ, ув-ию электрокинет-ого потенциала и к стабилизации сист за счет электрост-ого фактора;с др стороны, разб-ие ДС дисперсии-ой средой приведет к ум-ию ее вязкости и к нек-му сниж агрегативной уст-ти за счет гидродин-ого фактора. Опред защитного слоя: Доб к золю р-ра стаб повыш его агрег уст-ть.Коагул действ уменьш. Мин кол-во стаб, предотвр коагул 1 литра С(золя).В присутств порогового кол-ва электрол-коагул
43. Коагуляция.
Коагуляцией - процесс слипания -и добавлении к гидрозолю эл-та-коагулятора может происх:− сжатия диф-ого слоя ПИ (λ), что приводит к ум-ию потенциала на гран раздела адсорбц-го и диф-ого слоев ПИ (ϕδ) и электрокин-ого потенциала (ζ); − уменьш потенциала пов-сти (ϕ0), что приводит к ум-ию ϕδ и электрокин-кого потенциала (ζ)
Основными закономерностями: 1) коагулирующее действие оказывает тот ион электролита-коагулятора, знак заряда к-ого противоп знаку заряда коллоидной ч-цы;этот ион наз ионом-коагулятором (1-ое пр-ло Шульце–Гарди);
2) коагуляция гидрозолей начинается тогда, когда величина электрокинетического потенциала достигает критического значения (–30 мВ < ζ< 30 мВ (при T = 300 К));
3) величина порога коагуляции уменьшается при увеличении заряда иона-коагулятора, при этом пороги коагуляции гидрозоля под действием одно-, двух- и трехзарядных ионов-коагуляторов относятся как сотни, десятки и единицы (второе правило Шульце–Гарди):Na: Ca2: Al3 = 100:10:1 γ Cl:γSO4:γ PO43- = 100:10:1
|
|
|
4) коагул-щее действие ионов-коагуляторов одинакового заряда возрастает при ув-ии их ионного радиуса (т. е., в лиотропном ряду):γLi+ >γK+ >γCs+ происх это потому, что большие ионы гидратируются слабее маленьких, и их эф-ый заряд в ДСр (с учетомионной атмосферы) оказыв выше;
5) коагулирующая способность орг-ких ионов-коагуляторов выше, чем неорг-ских с таким же зарядом; причина этого заключ в том, что незаряженная часть орг-ских ионов явл гидрофобной,вследствие чего они интенсивнее адсорб-ся на пов-сти компактного агрегата и с большей легкостью нейтрализуют заряд ядра коллоидной частицы.
порог коагуляции в соответствии с теорией ДЛФО, понимают миним конц-ию электролита-коагулятора в ДС, при к-ой начинается коагуляция (медленная) гидрозоля (c м) Для осущ-ния быстрой коагуляции необх достичь в ДС такого значения конц-ции электролита-коагулятора, при к-ом (c б) скорость коагуляции достигает макс-ого значения и при дальнейшем ув-ии конц-ции электролита уже не изменяется. Величина порога коагуляции существенно зависит от момента его фиксирования, метода наблюдения за коагуляцией, а также от конц-ции гидрозоля.
На практике под порогом (медленной) коагуляции обычно понимают миним кол-во электролита, к-ое необходимо добавить к одному литру ДС (гидрозоля), чтобы за определ-ый промежуток врем можно было наблюдать видимый эффект коагуляции этого гидрозоля (помутнение, выпадение осадка, изменение окраски): γ= Сэл Vм/Vзоля
Под порогом быстрой коагуляции (γб) понимают миним кол-во электролита, после добавления к-ого к одному литру ДС скорость коагул достигает макс-ого значения и после добавл к ДС дополн порций эл-та уже не изм-ся.
Согласно второму пр-лу Шульце–Гарди, величина порога коагуляции при концентрац-ой коагуляции зависит от заряда иона-коагулятора несколько слабее, чем это предсказывает теория ДЛФО. Согласно теории ДЛФО, величина порога быстрой коагуляции гидрозоля в обл высоких потенциалов обратно пропорци-на заряду иона-коагулятора (z) в шестой, а в обл низких потенциалов – второй степени: γ= const/Z6
44. Теория ДЛФО (рассматрив процесс взаимод частиц ДФ по отдельн стадиям:перекрыв их пов-ных слоев и возникнов расклинив давления,исп понят потенц барьер, аналог энерг актив в химич кинетике.) – соотнош м-у силами притяж и отталк-ия ч-ц ДФ. Общая эн взаимод м-у ч-цами (U) в теории ДЛФО рассм-ся как сумма эн электростатич отталк (U отт) и мол-ного притяжения (U пр)
 U = U отт + U пр. Для сферич ч-ц ДФ эн отталк: Uотт=const ϕδ2exp(-h/λ) ϕδ – потенц на границе адсорб и диф слоев ПИ.
U = U отт + U пр. Для сферич ч-ц ДФ эн отталк: Uотт=const ϕδ2exp(-h/λ) ϕδ – потенц на границе адсорб и диф слоев ПИ.
|
|
|
Эн притяж двух сферич ч-ц зависит от расстоян м-у ними и м.б. рассчит: Uпр = -A r/12h2 А – константа мол-ных сил Гамакера. Чем сильнее взаимод-ют ДФ и ДСр, тем меньше величина A * и тем слабее силы притяжения м-у ч-цами ДФ.В соотв с теорией ДЛФО соотношения определяют поведение ДС, уст-сть (или скорость коагуляции) к-ых определяются знаком и величиной общей потенц эн взаимод ч-ц ДФ. Положит эн отталк U отт с ув-ем расст h уменьш по экспоненциальному з-ну, а отрицат эн притяж U пр обратно пропорц-на квадрату расстояния. В рез-те на малых расстояниях (при h → 0, U отт →const, U пр → –∞) и больших расстояниях (экспонента убывает значит-но быстрее, чем степенная ф-ция) м-у ч-цами преобладает эн притяжения, а на ср расстояниях – эн. электростат-ого отталк. Первичный минимум I отвечает непосредств-ому слипанию ч-ц ДФ, а вторичный минимум II – их притяжению через прослойки среды(формир-е соотв-но конденсац-но- кристалл-ных и коагуляционных стр-р). Отвечающий средн расстояниям максимум U * характеризует потенц барьер, препятствующий слипанию ч-ц. Ув-ию потенц-ого барьера способствует рост потенциала на пов-сти кол-ых ч-ц ϕδ в обл его малых значений. Эксперимент показывает, что уже при ⎟ϕδ⎟ ≈ 30 мВ возникает потенц барьер, обеспечивающий агрегативную уст-сть ДС. Следует отметить, что потенц барьер возрастает и при ув-ии константы Гамакера. Кривая а отвечает состоянию ДС с высоким потенц-ым барьером (U * >> kT) и неглубоким вторичным минимумом (U II < kT); в этом случае вероятность агрегирования ч-ц исчезающе мала и система явл-ся агрегативно уст-ой (нет коаг).Кривая б соотв-ет такому состоянию сист, при к-ом на любых расстояниях м-у ч-цами разность м-у эн притяж и отталк м-у ч-цами меньше тепловой эн (U * < kT, а также (U * + U II < kT)), вследствие чего сист явл-ся агрегативно неуст-ой и в ней происходит быстрая коагуляция с обр-ем плотных уст-ых агрегатов; в системах с жидкой или газообр ДФ наблюд коалесценция. Кривая в характеризуется наличием достаточно глубокого вторичного минимума (U II > kT или, хотя бы, U * + U II > kT); в такой сист наблюд интенсивное взаимод-ие ч-ц ДФ, к-ые, однако, не могут сблизиться на расстояния, отвечающие собств-о коагуляции, – рез-том такого взаимод явл структурирование сист, при к-ом все ч-цы ДФ находятся на определенном расстоянии друг от друга, взаим-уя Ч-з прослойки ДС (образуется гель).

Конденсац-о-кристаллизац-е структурообрз-ие, отвеч-ее
коагуляции в первичном (I) потенц-ом минимуме осущ-ся путем непосредств-ого хим-ого взаимод-ия м-у ч-цами ДФ и их срастания с обр-ем жесткой объемной стр-ры. Если ч-цы ДФ аморфны, то образующиеся стр-ры принято называть конденсационными, если кристаллич– то кристаллизац-ыми. ККС типичны для связнодисп-ых систем, т.е. систем с тв ДСр; эти стр-ры придают телам прочность, хрупкость и не восстанавливаются после разрушения. Под коагуляц-ми стр-ами понимают стр-ры, образующиеся при коагул во вторичном (II) минимум на потенц-ой кривой взаимод-ия ч-ц ДФ (хотя ККС также формируются в результате коагуляции). В этом случае взаимод-ие ч-ц ДФ осущ-ся ч-з прослойки ДСр, явл ван-дер-ваальс, поэтому простр-ые каркасы КС обычно непрочны. Механические св-ва таких стр-р определяются не столько св-вами ч-ц ДФ, сколько характером и особенн межчастичных взаимод и прослоек среды. КС обычно имеют жидкую ДСр, для них характерна способность восстанавливать стр-уру после ее разрушения; последнее явление носит назв тиксотропии.
Тиксотропия предст собой обратимое разрушение и послед восст-ие стр-ры ДС вследствие подвижности среды и броуновского дв-ия ч0ц ДФ. Восст-ие стр-ры приводит к ув-нию вязкости сист, поэтому Т -это уменьш вязкости сист во времени при приложении нагрузки и постепенный рост вязкости после снятия нагрузки. Чем медленнее восстанавливается структура после снятия нагрузки или чем медленнее она разрушается при определенном напряжении сдвига, тем сильнее для данной системы выражено явлении тиксотропии
ЭКСП метод опред порога коагул
1.В несколько пробирок налив по немного золя. В другие(такое же кол-во) пробирки наливают эл-т и воду. Потом в пробирку с золем приливают р-р электролита, включают секундомер, перемешивают смесь, переливая ее из пробирки в пробирку. После этого заполн кювету получ смесью и измеряют оптическую плотность ДС через одинаковый промежуток времени. Аналогично повторяется и с другими пробирками по увеличению концент. По получ данным строим график зав-ти D=f(vэл) опред Vэл=Vм, по формуле рассчит порог коагуляции: γ= Сэл Vм/Vзоля
В проц коагул происх слипание частиц золя в агрегаты, поэтому интенсивн света, рассеян сист, будет постеп увелич-ся.
Турбидиметрич метод.
Метод основ на измер интенсивн света, прошедш через сист, при услов что интенс-ть падающ свет потока ослабл в рез-те его рассеяния ДС.
Iпр=I0 – Iрас
46.Индифферетных электролитов.
Входящие в состав индифферентных эл-тов ионы не способны адсорбироваться на незаряженной пов-сти компактного агрегата СЕГ (иначе говоря, не способны образовывать осадки с ионами, входящими в состав компактного агрегата), вследствие чего они не входят в состав ядра коллоидной ч-цы гидрозоля и не могут влиять на величину потенциала поверхности ϕ0. Индифф-ые электролиты-коагуляторы способны вызывать только концентрационную коагуляцию гидрозолей. В зав-сти от величины заряда иона-коагулятора, вход-его в состав индифф-ого эл-та-коагулятора, возможны 2 случая.
В первом случае заряд иона-коагулятора не превышает (равен или меньше) по модулю величину заряда (ПОИ), входящих в состав исходного гидрозоля. СЕГ имеет следующий вид: {[AgCl] m n Cl– (n–x)K+} – x K+, тогда в кач электролитов-коагуляторов, удовлетворяющих указанному усл, могут выступать KNO3, NaF и любой др индифф-ый эл-лит с однозарядным катионом (для гидрозоля с отрицательно заряженной коллоидной ч-цей ионом-коагулятором явл-ся катион). Пусть мы добавляем к нашему гидрозолю KNO3. Т.к. нитрат калия по отношению к исходному гидрозолю является индифферентным, то величина ϕ0 гидрозоля не будет зависеть от конц-ции KNO3 в ДСр. Ув-ние конц-ции ПИ– ионов K+ – в ДС приведет к сжатию ДСПИ и уменьш электрокин-ого потенциала ζ, который при определенной конц-ции KNO3 в ДС (в изоэлектрической точке (ИЭТ)) станет равным нулю. Т.к. коагуляция гидрозоля начинается, когда ζ-потенциал достигает не нулевого, а нек-ого критического значения то на зав-сти потенциалов гидрозоля от конц-ции электролита-коагулятора в ДС появл зона коагуляции (ЗК), отвечающая условиям, когда ⎜ζ⎜< 30 мВ; при усл-ях ⎜ζ⎜> 30 мВ золь нах-ся в зоне уст-сти (ЗУ). В изоэлектрической точке СЕГ имеет след строение:{[AgCl] m n Cl– n K+}0. Образовавшийся в ЗК свежий осадок можно перевести во взвешенное состояние (обратно в золь) путем диссолюционной пептизации, просто добавляя в ДС воду; СЕГ после пептизации будет та же, что и до коагуляции когда заряд иона-коагулятора больше заряда ПОИ; для нашего гидрозоля электролитом, удовлетворяющим этому условию, явл-ся Al(NO3)3.В данном случае может происходить перезарядка (изменение знака) ζ (ϕ0=CONST) и на зав-сти потенциалов гидрозоля от конц-ии эл-та-коагулятора появл две зоны коагуляции и две зоны уст-сти (наблюд явление чередования зон уст-сти и коагуляции. В ИЭТ 1 и 2 стр-ие СЕГ выглядит след. обр:{[AgCl] mn Cl– n /3)Al3+}0 (ИЭТ 1), {[AgCl] m n Cl– (n /3+ y)Al3+ 3 y NO3–}0 (ИЭТ 2).Свежие осадки, образовавшиеся после коаг-ии в 1-ОЙ и второй зонах коагул,м.б.переведены во взвешенное состояние (обр. в гидрозоль),как и в предыд случае, при помощи диссолюц пептизации (добавление к ДС с осадком воды). После пептизации СЕГ будут им вид соотв-но: {[AgCl] m n Cl– (n /3– y)Al3+}3 y– y Al3+, {[AgCl] m n Cl– (n /3+ y)Al3+ (3 y–z)NO3–}z+ z NO3–
Неиндеферен
К неиндифф-ым относят эл-ты, ионы к-ых могут адсорбироваться на незаряженной пов-сти компактного агрегата ч-ц ДФ гидрозоля (способны образовывать осадки с ионами компактного агрегата), входя при этом в состав ядра кол-ой ч-цы и изменяя величину (в нек-ых случаях и знак) потенциала пов-сти ϕ0 и электрокин. потенциала ζ.
Неиндиф-ные эл-ты-коагуляторы способны вызывать нейтрализац-ую коагуляцию гидрозоля (в том случае, когда неиндиф-ым явл-ся ион-коагулятор). В зав-сти от знака заряда неиндиф-ого иона-коагулятора 2 случая:
 1. когда знак заряда неиндиф-ого иона совпадает со знаком ПОИ гидрозоля. Пусть у нас имеется отриц-но заряж гидрозоль AgCl, стр-рная ед-ца к-ого им вид: {[AgCl] m n Cl– (n–x)K+} – x K+, тогда в кач эл-та, удовлетв-их указанному усл, может выступать KCl или любой другой эл-т, содержащий анионы, способные образовывать осадки с ионами серебра. Поскольку пов-сть компактного агрегата(КА) исх-ого золя ненасыщена анионами Cl–,то при добавлении в ДC дополнит анионов хлора эти анионы способны адсорбир-ся на своб местах пов-сти; при этом происх «донасыщ» пов-сти КА ПОИ. После того, как хлорид-ионы заполнят все доступные для них места на пов-сти КА, осн. влияние на стр-ие СЕГ оказывают ионы калия (ионы-коагуляторы): ув-ие их конц-ции приводит к сжатию ДСПИ, переходу всех ПИ в адсорбц-ый слой и ум-ию ζ (в ИЭТ – до нулевого значения).Стр-ие СЕГ в ИЭТ им вид: {[AgCl] m (n+x)Cl– (n+x)K+}0.свежий осадок, образовавшийся в процессе коагуляции, м.б. переведен обратно в гидрозоль путем диссолюционной пептизации, причем СЕГ после пептизации {[AgCl] m (n+x)Cl– (n+x–z)K+}z+ z K+.
1. когда знак заряда неиндиф-ого иона совпадает со знаком ПОИ гидрозоля. Пусть у нас имеется отриц-но заряж гидрозоль AgCl, стр-рная ед-ца к-ого им вид: {[AgCl] m n Cl– (n–x)K+} – x K+, тогда в кач эл-та, удовлетв-их указанному усл, может выступать KCl или любой другой эл-т, содержащий анионы, способные образовывать осадки с ионами серебра. Поскольку пов-сть компактного агрегата(КА) исх-ого золя ненасыщена анионами Cl–,то при добавлении в ДC дополнит анионов хлора эти анионы способны адсорбир-ся на своб местах пов-сти; при этом происх «донасыщ» пов-сти КА ПОИ. После того, как хлорид-ионы заполнят все доступные для них места на пов-сти КА, осн. влияние на стр-ие СЕГ оказывают ионы калия (ионы-коагуляторы): ув-ие их конц-ции приводит к сжатию ДСПИ, переходу всех ПИ в адсорбц-ый слой и ум-ию ζ (в ИЭТ – до нулевого значения).Стр-ие СЕГ в ИЭТ им вид: {[AgCl] m (n+x)Cl– (n+x)K+}0.свежий осадок, образовавшийся в процессе коагуляции, м.б. переведен обратно в гидрозоль путем диссолюционной пептизации, причем СЕГ после пептизации {[AgCl] m (n+x)Cl– (n+x–z)K+}z+ z K+.
 2. знак заряда неидифф иона противоположен знаку заряда ПОИ. Для исходного гидрозоля в кач неиндифф-ого эл-та, удовлетворяющего этому условию, может выступать AgNO3 (или любой эл-т, содержащий катионы, способные образовывать осадок с ионами хлора). При добавлении к гидрозолю AgCl с отрицат заряж-ыми кол-ыми ч-цами AgNO3 неиндиф-ые ионы Ag+ связывают ПОИ Cl– в ТР соединение AgCl, нейтрализуют потенциал пов-сти; при этом происх нейтрализац-ая коагуляция гидрозоля. При добавлении к ДС дополнительных порций AgNO3 ионы серебра продолжают адсорбироваться на пов-сти КА, вследствие чего кол-ая ч-ца приобретает положит заряд При дальнейшем повышении конц-ции AgNO3 в ДС происходит концентрац-ая коагуляции гидрозоля за счет сжатия ДСПИ. В ИЭТ 1 и 2 стр-е СЕГ выглядит след образом:{[AgCl] m+n }0 (ИЭТ 1), {[AgCl] m+n k Ag+ k NO3–}0 (ИЭТ 2).
2. знак заряда неидифф иона противоположен знаку заряда ПОИ. Для исходного гидрозоля в кач неиндифф-ого эл-та, удовлетворяющего этому условию, может выступать AgNO3 (или любой эл-т, содержащий катионы, способные образовывать осадок с ионами хлора). При добавлении к гидрозолю AgCl с отрицат заряж-ыми кол-ыми ч-цами AgNO3 неиндиф-ые ионы Ag+ связывают ПОИ Cl– в ТР соединение AgCl, нейтрализуют потенциал пов-сти; при этом происх нейтрализац-ая коагуляция гидрозоля. При добавлении к ДС дополнительных порций AgNO3 ионы серебра продолжают адсорбироваться на пов-сти КА, вследствие чего кол-ая ч-ца приобретает положит заряд При дальнейшем повышении конц-ции AgNO3 в ДС происходит концентрац-ая коагуляции гидрозоля за счет сжатия ДСПИ. В ИЭТ 1 и 2 стр-е СЕГ выглядит след образом:{[AgCl] m+n }0 (ИЭТ 1), {[AgCl] m+n k Ag+ k NO3–}0 (ИЭТ 2).
Осадок, полученный после коагуляции в первой ЗК, можно перевести во взвешенное состояние путем адсорбционной пептизации.При этом, если к осадку добавлять эл-т KCl, то СЕГ будет им. Вид {[AgCl] m+n k Cl– (k – z)K+} z – z K+, а если AgNO3, то{[AgCl] m+n k Ag+ (k – z)NO3–}z+ z NO3–.
Виды коагуляции
различают концентрац-ную и нейтрализац-ую коагул.
Концентрац-ая коаг вызывается ув-ем конц-ции ПИ в ДСр и в АСПИ, что приводит к сжатию ДСПИ. Этот вид коаг-ии характерен для ДС с высоким потенциалом пов-сти.
Концентрационная коагуляция гидрозолей протекает при доб к ним индифф-ых эл-тов-коагуляторов, т. е. эл-тов, ионы к-ых не способны адсорбироваться на пов-сти КА гидрозоля и нейтрализовать его пов-ый потенциал (ϕ0). Напр, для гидрозоля {[AgCl] m n Cl– (n – x)K+}x– x K+. такими эл-тами явл-ся, напр, KNO3 и NaF. Нейтрализационная коагуляция просх вследствие ум-ния (нейтрализации) потенциала пов-сти (ϕ0), что происх в рез-те связывания ПОИ в ТР соед-ие. Этот вид коагуляции характерен для ДС с низкими значениями потенциала поверхности.
 Нейтрализационная коагуляция гидрозолей протекает при добавлении к ним неиндифф-ых эл-тов, т. е. электролитов, ионы к-ых способны адсорбироваться на пов-сти КА ч-цы ДФ (кроме того, неиндиф-ые ионы этих электролитов должны им заряд, противопол заряду ПОИ гидрозоля). Для гидрозоля, схема стр-рной ед-цы к-ого приведена выше, такими эл-тами явл-ся, например, AgNO3 или Pb(NO3)2. Под скоростью коагуляции (ϑ) понимают изменение частичной конц-ции ДФ (ν) в ед-цу времени ϑ =− d ν/ dt. Обл I – система агрегативно уст-ва, т.к. потенциальный барьер высок (U * >> kT), и ч-цы ДФ не могут его преодолеть; ϑ ≈ 0, но, поскольку отдельные частицы ДФ могут, сталкиваясь, слипаться, область называют «областью скрытой коагуляции».
Нейтрализационная коагуляция гидрозолей протекает при добавлении к ним неиндифф-ых эл-тов, т. е. электролитов, ионы к-ых способны адсорбироваться на пов-сти КА ч-цы ДФ (кроме того, неиндиф-ые ионы этих электролитов должны им заряд, противопол заряду ПОИ гидрозоля). Для гидрозоля, схема стр-рной ед-цы к-ого приведена выше, такими эл-тами явл-ся, например, AgNO3 или Pb(NO3)2. Под скоростью коагуляции (ϑ) понимают изменение частичной конц-ции ДФ (ν) в ед-цу времени ϑ =− d ν/ dt. Обл I – система агрегативно уст-ва, т.к. потенциальный барьер высок (U * >> kT), и ч-цы ДФ не могут его преодолеть; ϑ ≈ 0, но, поскольку отдельные частицы ДФ могут, сталкиваясь, слипаться, область называют «областью скрытой коагуляции».
Обл II – «область медленной коагуляции» (система становится агрегативно неуст-вой); при конц-ции эл-та, отвечающей порогу медленной коагуляции (γм) потенц барьер (U *) и электрокин потенциал (ζ) ч-ц ДФ снижается настолько, что наиболее быстрые ч-цы оказываются в состоянии преодолеть потенц барьер и скоагулировать; с ростом конц-ции электролита-коагулятора потенц барьер и ζ-потенциал сниж-ся, а скорость коаг возрастает (ϑ = f (c)).
Обл III – «область быстрой коагуляции»; при достижении конц-ции эл-та, отвечающей порогу быстрой коагуляции (γб) потенц барьер снижается настолько (U * ≈ 0), что любое столкн-ие ч-ц ДФ приводит к их слипанию (коаг-ии); скорость коагуляции достигает максим-ого значения и уже не изм-ся при ув-ии конц-ции эл-та-коагулятора (ϑ ≠ f (c)).
Теория быстрой коагуляции столкновение ч-ц ДФ явл-ся не только необходимым, но и достаточным условием коагуляции (любое столкн-ие ч-ц ДФ приводит к коагуляции); учитываются только парные столкновения ч-ц ДФ, как наиб вероятные. Т.о., кинетику быстрой коагуляции можно описывать как реакцию второго порядка: скорость коагуляции пропорц-на квадрату частичной концентрации частиц ДФ в соответствии с уравнением ϑ = k к ν2
 характеристическое время, или время половинной коагуляции (τ1/2)
характеристическое время, или время половинной коагуляции (τ1/2)
ур-ие Гельмгольца–Смолуховского
для быстрой каогуляции 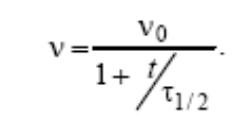
49 НЕйтрализ и концентрац.
Концентрационная коагуляция вызывается увеличением концентрации противоионов в дисперсионной среде и в адсорбционном слое противоионов, что приводит к сжатию диффузного слоя противоионов. Данный вид коагуляции характерен для дисперсных систем с высоким потенциалом поверхности.
Концентрационная коагуляция гидрозолей протекает при доб к ним индифферентных электролитов-коагуляторов, т. е. электролитов, ионы которых не способны адсорбироваться на поверхности компактного агрегата гидрозоля и нейтрализовать его поверхностный потенциал (ϕ0). Например, для гидрозоля
{[AgCl] m n Cl– (n – x)K+}x– x K+.такими электролитами являются, например, KNO3 и NaF. Нейтрализационная коагуляция просходит вследствие уменьшения (нейтрализации) потенциала поверхности (ϕ0), что происходит в результате связывания потенциалопределяющих ионов в труднорастворимое соединение. Этот вид коагуляции характерен для ДС с низкими значениями потенциала поверхности.
Нейтрализационная коагуляция гидрозолей протекает при добавлении к ним неиндифферентных электролитов, т. е. электролитов, ионы которых способны адсорбироваться на поверхности КА частицы ДФ (кроме того, неиндифферентные ионы этих электролитов должны иметь заряд, противоположный заряду ПОИ гидрозоля). Для гидрозоля, схема структурной единицы которого приведена выше, такими электролитами являются, например, AgNO3 или Pb(NO3)2.
Флокуляция
Флокуляция – потеря уст-сти под действием высокомолек соедин(ВМС) называют флокуляцией, а эти ВМС – флокулянтами При флок теряется агрегатив,затем седимент уст-ть.образ рыхлые хлопьевидн стр-ры – флокулы(неорг полимеры, орган высокомол соедин)синтетич(неионоген и полиэлектролиты(анион и катионные)) к-ые в зав-сти от соотношения плотностей в-ва ДФ и ДСр оседают либо всплывают. Силы,выз адсорбц ВМС - силы межмол-ного притяжения (дисперсионные, индукционные и ориентационные), а также химические силы (водородная, ионная, ковалентная связь).
Протек по 2 механизмам: 1.нейтрализационный:осущ действ макроионов полимера,имеющ заряд, противопол по знаку заряду ч-цы(макроионы катион. флокулянтов адсорбир на отриц заряж участках,а макроионы анион флокулянтов – на положит) 2.мостичный – вначале происх адсорб макромол-л на одной частице ДФ.мол флок закрепляется частью своих сегментов на пов-ти частиц. Не связ с данной частиц сегменты макромол-л закрепл на свободн пов-ти близлежащ ч-ц обр мостики м-у ч-цами ДФ.
Наличием данного механизма подтвержд: флокуляция ч-ц ДС как заряженными, так и незаряженными флокулянтами; обр-ие в присутствии флокулянтов более объемных и рыхлых осадков, чем при коагуляции под действием электролитов; рост флокулирующего действия флокулянтов с ув-ем их мол-рной массы; возможность флокуляции заряж-ых ч-ц одноименно заряж полиионами.
Седиментац метод:Сущ-ет:прямые и косв-ые.прямые- агрегаты ч-ц в ДС.(оптич методы). Косвен- изуч не сам проц,а его влияние на опред св-ва ДС(электро-пров,диэлектр прониц и др)степень флок.
Суть метода:измерение через опред промежутов времени объема осадка,обр в рез-те оседания частиц.по эксп данным рассчит объем осветл части ДС в кажд момент времени от ачал порц седиментации: Vосв=Vобщ-Vосад
Опред степень осветления суспензии:α=Vосв/Vобщ
Строим граф зав-ть α=f(t),опред осветл суспензии, для нашего α как в отсутсвии t0, Так и в присутвии tп полимера-флокулянта. Рассчитываем параметр флокулир действия полимера:
Дα= t0 /tп
если Дα>0, то полимер – флокулянт, если наоборот, то он стабилизирует ДС.
Флокулир действие тем больше,чем больше значение Д
Оптические св-ва
ДС явл-ся оптич неоднор.Их оптич св-ва обусл взаимод ЭМИ, облад опред энергией,с частиц ДФ и мол ДСр. При прохожд свет волны через ДС набл явл: поглощения, рассеян, отраж,преломления. Преломл – происх на границе ДС и ОС,а также внутри ДС на гр раздела фаз ДФ/ДСр.
В зав-ти от соотнош длины волны и диаметра ч-цы ДФ,дисп сист деляться на: d>λ-грубодисп,d<λ – высокодисперсные. d>λ лучи света попад на пов-ть частиц,отраж и преломл.ПОГЛОЩЕНИЕ опред природ в-в и не зав от размера ч-ц; d<λ для них хар-но рассеяние.
I пр = I 0(пад) − I рас − I отр – I погл. Зав-сть I пр от толщины слоя ДС, через к-ый проходит свет (l), выражается ф-лой: I пр = I 0 exp(− kl), k – экстинция, или коэф-т ослабления светового потока,к-ый определяется разл оптич явлениями
Хар-ка,оценив ослабл интенсивн пад светов потока- оптич плотность: D=lg(Io/Iпр), коэф пропуск: T= Iпр/Io D= -lgT
При пропуск свет потока через золь каждая ч-ца ДФ рассеив свет во всех напр
 Теория светорассеяния ( Рэлеем) для разбавленных оптически неоднор (n ДФ ≠ n ДСр) ДС, содержащих сферич, не поглощающие свет и не проводящие электрич ток ч-цы, диаметр к-ых одинаков и составляет меньше одной десятой длины волны падающего света (d ≤ 0,1λ)
Теория светорассеяния ( Рэлеем) для разбавленных оптически неоднор (n ДФ ≠ n ДСр) ДС, содержащих сферич, не поглощающие свет и не проводящие электрич ток ч-цы, диаметр к-ых одинаков и составляет меньше одной десятой длины волны падающего света (d ≤ 0,1λ)
Ур-ние Рэлея

Если рассм под углом 90 по отн к пад свету,то когда n ДФ ≈ n ДСр, явл-ие светорас-ия в ДС отсутствует. мутность сист (τ), к-ая показ-ет долю света, рас-го ч-цами ДФ по отнош к инт-сти падающего на сист света: τ=Iрас/Iо
Если свет, прошедший через ДС только рассеивается, а др оптич явления отсутствуют, то в ур-ии k' = τ, и оно им вид
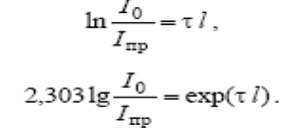
 |

Подставим в ур-ние Рэлея,получим:

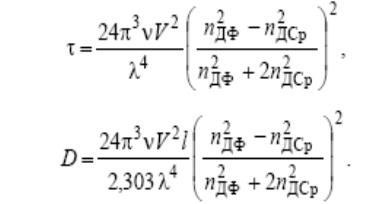
оптические хар-ки ДС с размерами ч-ц d ≤ 0,1λ обр пропорц-ны длине волны в 4 степени. Экспериментально было установлено,что с ув-ем
размера ч-ц зав-сть не соблюдается и им. место соотношение(Геллера): D=K/λm , m – показатель дисп-сти системы (1 ≤ m ≤ 4 для систем, размеры ч-ц ДФ к-ых наход в пределах 0,1λ ≤ d ≤ 0,3λ). ДС, для к-ых выполн соотнош Рэлея и Геллера, иногда наз «белыми золями», поскольку эти ДС явл-ся высокодисперсными и не поглощают свет в видимой области спектра. В нефелометрич и турбидим-ом методах исслед определяют свва ДС в целом.
|
|
|
