 |


Влияние температуры на положение равновесия
|
|
|
|
Повышение либо понижение температуры означает приобретение либо потерю системой энергии и, следовательно, должно изменять величину константы равновесия.
Запишем уравнение (I.99) в следующем виде:
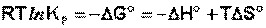 (I.104)
(I.104)
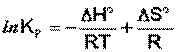 (I.105)
(I.105)
Продифференцировав выражение (I.105) по температуре, получаем для зависимости константы равновесия от температуры – изобару Вант-Гоффа:
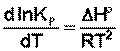 (I.06)
(I.06)
Изобара Вант-Гоффа связывает изменение константы химического равновесия с тепловым эффектом реакции в изобарных условиях. Очевидно, что чем больше по абсолютной величине тепловой эффект химической реакции, тем сильнее влияет температура на величину константы равновесия. Если реакция не сопровождается тепловым эффектом, то константа равновесия не зависит от температуры.
Экзотермические реакции: ΔH° < 0. В этом случае, согласно (I.106), температурный коэффициент логарифма константы равновесия отрицателен. Повышение температуры уменьшает величину константы равновесия, т.е. смещает равновесие влево.
Эндотермические реакции: ΔH° > 0. В этом случае температурный коэффициент логарифма константы равновесия положителен; повышение температуры увеличивает величину константы равновесия (смещает равновесие вправо).
Графики зависимостей константы равновесия от температуры для экзотермических и эндотермических реакций приведены на рис. I.4.
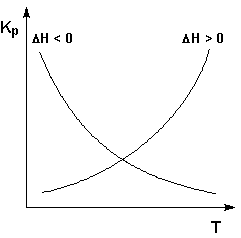
Рис. 1.4. Зависимость константы равновесия от температуры
Действие рассмотренных факторов (давления, концентрации и температуры), равно как и любых других, на систему, находящуюся в состоянии равновесия, обобщает принцип смещения равновесия, называемый также принципом Ле Шателье – Брауна:
Если на систему, находящуюся в состоянии истинного равновесия, оказывается внешнее воздействие, то в системе возникает самопроизвольный процесс, компенсирующий данное воздействие.
|
|
|
Принцип Ле Шателье – Брауна является одним из следствий второго начала термодинамики и применим к любым макроскопическим системам, находящимся в состоянии истинного равновесия.
ФАЗОВЫЕ РАВНОВЕСИЯ
Вещество при изменении давления и температуры может переходить из одного агрегатного состояния в другое. Эти переходы, совершающиеся при постоянной температуре, называют фазовыми переходами первого рода. Количество теплоты, которое вещество получает из окружающей среды либо отдает окружающей среде при фазовом переходе, есть скрытая теплота фазового перехода λ фп. Если рассматривается гетерогенная система, в которой нет химических взаимодействий, а возможны лишь фазовые переходы, то при постоянстве температуры и давления в системе существует т.н. фазовое равновесие. Фазовое равновесие характеризуется некоторым числом фаз, компонентов и числом степеней термодинамической свободы системы.
Компонент – химически однородная составная часть системы, которая может быть выделена из системы и существовать вне её. Число независимых компонентов системы равно числу компонентов минус число возможных химических реакций между ними.
Число степеней свободы – число параметров состояния системы, которые могут быть одновременно произвольно изменены в некоторых пределах без изменения числа и природы фаз в системе.
Число степеней свободы гетерогенной термодинамической системы, находящейся в состоянии фазового равновесия, определяется правилом фаз, сформулированным Дж. Гиббсом:
Число степеней свободы равновесной термодинамической системы С равно числу независимых компонентов системы К минус число фаз Ф плюс число внешних факторов, влияющих на равновесие.
Для системы, на которую из внешних факторов влияют только температура и давление, можно записать:
|
|
|
С = К – Ф + 2 (I.108)
Системы принято классифицировать по числу компонентов (одно-, двухкомпонентные и т.д.), по числу фаз (одно-, двухфазные и т.д.) и числу степеней свободы (инвариантные, моно-, дивариантные и т.д.). Анализ диаграмм состояния позволяет определить число фаз в системе, границы их существования, характер взаимодействия компонентов. В основе анализа диаграмм состояния лежат два принципа: принцип непрерывности и принцип соответствия. Согласно принципу непрерывности, при непрерывном изменении параметров состояния все свойства отдельных фаз изменяются также непрерывно; свойства системы в целом изменяются непрерывно до тех пор, пока не изменится число или природа фаз в системе, что приводит к скачкообразному изменению свойств системы. Согласно принципу соответствия, на диаграмме состояния системы каждой фазе соответствует часть плоскости – поле фазы. Линии пересечения плоскостей отвечают равновесию между двумя фазами. Всякая точка на диаграмме состояния (т.н. фигуративная точка) отвечает некоторому состоянию системы с определенными значениями параметров состояния.
Рассмотрим и проанализируем диаграмму состояния воды. Поскольку вода – единственное присутствующее в системе вещество, число независимых компонентов К = 1. В системе возможны три фазовых равновесия: между жидкостью и газом (линия ОА – зависимость давления насыщенного пара воды от температуры), твердым телом и газом (линия ОВ – зависимость давления насыщенного пара надо льдом от температуры), твердым телом и жидкостью (линия ОС – зависимость температуры плавления льда от давления). Три кривые имеют точку пересечения О, называемую тройной точкой воды; тройная точка отвечает равновесию между тремя фазами.
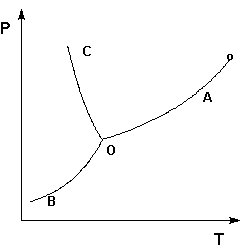 Диаграмма состояния воды
Диаграмма состояния воды
В тройной точке система трехфазна и число степеней свободы равно нулю; три фазы могут находиться в равновесии лишь при строго определенных значениях температуры и давления (для воды тройная точка отвечает состоянию с Р = 6.1 кПа и Т = 273.16 К).
Кривая ОВ теоретически продолжается до абсолютного нуля, а кривая давления насыщенного пара над жидкостью ОА заканчивается в критической точке воды (Tкр = 607.46 К, Ркр = 19.5 МПа); выше критической температуры газ и жидкость не могут существовать как отдельные фазы. Кривая ОС в верхней части (при высоких давлениях) изменяет свой наклон (появляются новые кристаллические фазы, плотность которых, в отличие от обычного льда, выше, чем у воды).
|
|
|
Внутри каждой из областей диаграммы (АОВ, ВОС, АОС) система однофазна; число степеней свободы системы равно двум (система дивариантна), т.е. можно одновременно изменять и температуру, и давление, не вызывая изменения числа фаз в системе:
С = 1 – 1 + 2 = 2
На каждой из линий число фаз в системе равно двум и, согласно правилу фаз, система моновариантна, т.е. для каждого значения температуры имеется только одно значение давления, при котором система двухфазна:
С = 1 – 2 + 2 = 1
Влияние давления на температуру фазового перехода описывает уравнение Клаузиуса – Клапейрона:
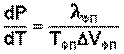 (I.109)
(I.109)
Здесь ΔVфп = V2 – V1 есть изменение молярного объема вещества при фазовом переходе (причем V2 относится к состоянию, переход в которое сопровождается поглощением теплоты). Уравнение Клаузиуса – Клапейрона позволяет объяснить наклон кривых равновесия на диаграмме состояния однокомпонентной системы. Для переходов "жидкость – пар" и "твердое вещество – пар" ΔV всегда больше нуля; поэтому кривые на диаграмме состояния, отвечающие этим равновесиям, всегда наклонены вправо (повышение температуры всегда увеличивает давление насыщенного пара). Поскольку молярный объем газа много больше молярного объема того же вещества в жидком или твердом агрегатном состояниях (Vг >> Vж, Vг >> Vт), уравнение (I.109) для частных случаев испарения и возгонки примет следующий вид:
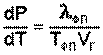 (I.110)
(I.110)
Для многих веществ скрытая теплота парообразования или возгонки постоянна в большом интервале температур; в этом случае уравнение (I.110) можно проинтегрировать:
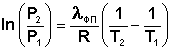 (I.111)
(I.111)
Кривая равновесия "твердое вещество – жидкость" на диаграммах состояния воды и висмута наклонена влево, а на диаграммах состояния остальных веществ – вправо. Это связано с тем, что плотность воды больше, чем плотность льда (и плотность жидкого висмута больше его плотности в твердом состоянии), т.е. плавление сопровождается уменьшением объема (ΔV < 0). Как следует из выражения (I.111), в этом случае увеличение давления будет понижать температуру фазового перехода "твердое тело – жидкость" (воду и висмут относят поэтому к т.н. аномальным веществам). Для всех остальных веществ (т.н. нормальные вещества) ΔVпл > 0 и, согласно уравнению Клаузиуса – Клапейрона, увеличение давления приводит к повышению температуры плавления.
|
|
|
ХИМИЧЕСКАЯ КИНЕТИКА
Законы химической термодинамики позволяют определить направление и предел протекания возможного при данных условиях химического процесса, а также его энергетический эффект. Однако термодинамика не может ответить на вопросы о том, как осуществляется данный процесс и с какой скоростью. Эти вопросы – механизм и скорость химической реакции – и являются предметом химической кинетики.
СКОРОСТЬ ХИМИЧЕСКОЙ РЕАКЦИИ
Скорость химической реакции есть число элементарных актов химической реакции, происходящих в единицу времени в единице объема (для гомогенных реакций) или на единице поверхности (для гетерогенных реакций).
Скорость химической реакции есть изменение концентрации реагирующих веществ в единицу времени.
Скорость реакции считается положительной, поэтому математически определение средней скорости реакции в интервале времени Δt записывается следующим образом:
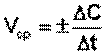 (II.1)
(II.1)
Графическое изображение зависимости концентрации реагентов от времени есть кинетическая кривая
 Кинетические кривые для исходных веществ (А) и продуктов реакции (В).
Кинетические кривые для исходных веществ (А) и продуктов реакции (В).
В том случае, если стехиометрические коэффициенты в уравнении химической реакции неодинаковы, величина скорости реакции будет зависеть от того, изменение концентрации какого реагента определялось.
Скорость химической реакции зависит от множества факторов: природы реагирующих веществ, их концентрации, температуры, природы растворителя и т.д.
Кинетическое уравнение химической реакции. Порядок реакции
Одной из задач, стоящих перед химической кинетикой, является определение состава реакционной смеси (т.е. концентраций всех реагентов) в любой момент времени, для чего необходимо знать зависимость скорости реакции от концентраций. В общем случае, чем больше концентрации реагирующих веществ, тем больше скорость химической реакции. В основе химической кинетики лежит т.н. основной постулат химической кинетики:
Скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ, взятых в некоторых степенях.
|
|
|
Т. е. для реакции
аА + bВ + dD +... ––> еЕ +...
можно записать:
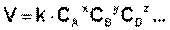 (II.4)
(II.4)
где k есть константа скорости химической реакции. Константа скорости численно равна скорости реакции при концентрациях всех реагирующих веществ, равных 1 моль/л.
Зависимость скорости реакции от концентраций реагирующих веществ определяется экспериментально и называется кинетическим уравнением химической реакции. Показатель степени при концентрации каждого из реагирующих веществ в кинетическом уравнении химической реакции (в уравнении (II.4) соответственно x, y и z) есть частный порядок реакции по данному компоненту. Сумма показателей степени в кинетическом уравнении химической реакции (x + y + z) представляет собой общий порядок реакции.
В химической кинетике принято классифицировать реакции по величине общего порядка реакции. Рассмотрим зависимость концентрации реагирующих веществ от времени для необратимых (односторонних) реакций нулевого, первого и второго порядков.
Реакции нулевого порядка
Для реакций нулевого порядка кинетическое уравнение имеет следующий вид:
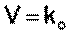 (II.5)
(II.5)
Скорость реакции нулевого порядка постоянна во времени и не зависит от концентраций реагирующих веществ; это характерно для многих гетерогенных (идущих на поверхности раздела фаз) реакций в том случае, когда скорость диффузии реагентов к поверхности меньше скорости их химического превращения.
Реакции первого порядка
Рассмотрим зависимость от времени концентрации исходного вещества А для случая реакции первого порядка А ––> В. Реакции первого порядка характеризуются кинетическим уравнением следующего вида (II.6).
 (II.7)
(II.7)
После интегрирования выражения (II.7) получаем:
 (II.8)
(II.8)
Константу интегрирования g определим из начальных условий: в момент времени t = 0 концентрация С равна начальной концентрации Со. Отсюда следует, что g = ln Со. Получаем:
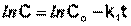 (II.9)
(II.9)
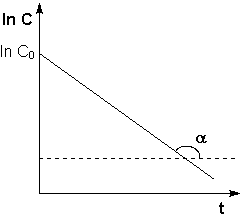
Зависимость логарифма концентрации от времени для реакций первого порядка
Т.о., логарифм концентрации для реакции первого порядка линейно зависит от времени и константа скорости численно равна тангенсу угла наклона прямой к оси времени.
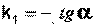 (II.10)
(II.10)
Из уравнения (II.9) легко получить выражение для константы скорости односторонней реакции первого порядка:
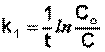 (II.11)
(II.11)
Еще одной кинетической характеристикой реакции является период полупревращения t1/2 – время, за которое концентрация исходного вещества уменьшается вдвое по сравнению с исходной. Выразим t1/2 для реакции первого порядка, учитывая, что С = ½Со:
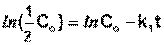 (II.12)
(II.12)
Отсюда
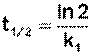 (II.13)
(II.13)
Отсюда видно, что период полупревращения реакции первого порядка не зависит от начальной концентрации исходного вещества.
Реакции второго порядка
Для реакций второго порядка кинетическое уравнение имеет следующий вид:
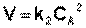 (II.14)
(II.14)
либо
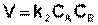 (II.15)
(II.15)
Рассмотрим простейший случай, когда концентрации исходных веществ одинаковы; уравнение (II.14) в этом случае можно переписать следующим образом:
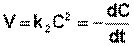 (II.16)
(II.16)
После разделения переменных и интегрирования получаем:
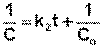 (II.18)
(II.18)
Т.о., для реакций второго порядка, имеющих кинетическое уравнение вида (II.14), характерна линейная зависимость обратной концентрации от времени (рис. 2.4) и константа скорости равна тангенсу угла наклона прямой к оси времени:
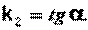 (II.19)
(II.19)
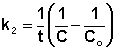 (II.20)
(II.20)
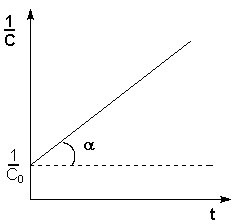
Зависимость обратной концентрации от времени для реакций второго порядка
Порядок химической реакции есть формально-кинетическое понятие, физический смысл которого для элементарных (одностадийных) реакций заключается в следующем: порядок реакции равен числу одновременно изменяющихся концентраций. В случае элементарных реакций порядок реакции может быть равен сумме коэффициентов в стехиометрическом уравнении реакции; однако в общем случае порядок реакции определяется только из экспериментальных данных и зависит от условий проведения реакции.
Молекулярность элементарных реакций
Элементарными (простыми) называют реакции, идущие в одну стадию. Их принято классифицировать по молекулярности:
Молекулярность элементарной реакции – число частиц, которые, согласно экспериментально установленному механизму реакции, участвуют в элементарном акте химического взаимодействия.
Мономолекулярные – реакции, в которых происходит химическое превращение одной молекулы (изомеризация, диссоциация и т. д.):
I2 ––> I• + I•
Бимолекулярные – реакции, элементарный акт которых осуществляется при столкновении двух частиц (одинаковых или различных):
СН3Вr + КОН ––> СН3ОН + КВr
Тримолекулярные – реакции, элементарный акт которых осуществляется при столкновении трех частиц:
О2 + NО + NО ––> 2NО2
Реакции с молекулярностью более трёх неизвестны.
Для элементарных реакций, проводимых при близких концентрациях исходных веществ, величины молекулярности и порядка реакции совпадают. Тем не менее, никакой чётко определенной взаимосвязи между понятиями молекулярности и порядка реакции не существует.
Сложные реакции
Сложными называют химические реакции, протекающие более чем в одну стадию. Рассмотрим в качестве примера одну из сложных реакций, кинетика и механизм которой хорошо изучены:
2НI + Н2О2 ––> I2 + 2Н2О
Данная реакция является реакцией второго порядка; её кинетическое уравнение имеет следующий вид:
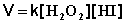 (II.28)
(II.28)
Изучение механизма реакции показало, что она является двухстадийной (протекает в две стадии):
1) НI + Н2О2 ––> НIО + Н2О
2) НIО + НI ––> I2 + Н2О
Скорость первой стадии V1 много больше скорости второй стадии V2 и общая скорость реакции определяется скоростью более медленной стадии, называемой поэтому скоростьопределяющей или лимитирующей.
Классификация сложных реакций
Последовательные реакции
Последовательными называются сложные реакции, протекающие таким образом, что вещества, образующиеся в результате одной стадии (т.е. продукты этой стадии), являются исходными веществами для другой стадии. Схематически последовательную реакцию можно изобразить следующим образом:
А ––> В ––> С ––>...
Параллельные реакции.
Параллельными называют химические реакции, в которых одни и те же исходные вещества одновременно могут образовывать различные продукты реакции, например, два или более изомера:
Сопряжённые реакции.
Сопряжёнными принято называть сложные реакции, протекающие следующим образом:
1) А + В ––> С
2) А + D ––> Е,
причём одна из реакций может протекать самостоятельно, а вторая возможна только при наличии первой. Вещество А, общее для обеих реакций, носит название актор, вещество В – индуктор, вещество D, взаимодействующее с А только при наличии первой реакции – акцептор.
Цепные реакции
Цепными называют реакции, состоящие из ряда взаимосвязанных стадий, когда частицы, образующиеся в результате каждой стадии, генерируют последующие стадии. Как правило, цепные реакции протекают с участием свободных радикалов. Для всех цепных реакций характерны три типичные стадии, которые рассмотрим на примере фотохимической реакции образования хлороводорода.
1. Зарождение цепи (инициация):
Сl2 + hν ––> 2 Сl•
2. Развитие цепи:
Н2 + Сl• ––> НСl + Н•
Н• + Сl2 ––> НСl + Сl•
Стадия развития цепи характеризуется числом молекул продукта реакции, приходящихся на одну активную частицу – длиной цепи.
3. Обрыв цепи (рекомбинация):
Н• + Н• ––> Н2
Сl• + Сl• ––> Сl2
Н• + Сl• ––> НСl
Обрыв цепи возможен также при взаимодействии активных частиц с материалом стенки сосуда, в котором проводится реакция, поэтому скорость цепных реакций может зависеть от материала и даже от формы реакционного сосуда.
Реакция образования хлороводорода является примером неразветвленной цепной реакции – реакции, в которой на одну прореагировавшую активную частицу приходится не более одной вновь возникающей. Разветвленными называют цепные реакции, в которых на каждую прореагировавшую активную частицу приходится более одной вновь возникающей, т.е. число активных частиц в ходе реакции постоянно возрастает.
Влияние температуры на константу скорости реакции
Константа скорости реакции есть функция от температуры; повышение температуры, как правило, увеличивает константу скорости. Первая попытка учесть влияние температуры была сделана Я. Г. Вант-Гоффом, который сформулировал следующее эмпирическое правило:
При повышении температуры на каждые 10 градусов константа скорости элементарной химической реакции увеличивается в 2 – 4 раза.
Величина, показывающая, во сколько раз увеличивается константа скорости при повышении температуры на 10 градусов, есть температурный коэффициент константы скорости реакции γ. Математически правило Вант-Гоффа можно записать следующим образом:
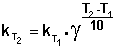 (II.30)
(II.30)
Правило Вант-Гоффа применимо лишь в узком температурном интервале, поскольку температурный коэффициент скорости реакции γ сам является функцией от температуры; при очень высоких и очень низких температурах γ становится равным единице (т.е. скорость химической реакции перестает зависеть от температуры).
Уравнение Аррениуса
Взаимодействие частиц осуществляется при их столкновениях; однако число столкновений молекул очень велико и, если бы каждое столкновение приводило к химическому взаимодействию частиц, все реакции протекали бы практически мгновенно. С. Аррениус постулировал, что столкновения молекул будут эффективны (т.е. будут приводить к реакции) только в том случае, если сталкивающиеся молекулы обладают некоторым запасом энергии – энергией активации.
Энергия активации есть минимальная энергия, которой должны обладать молекулы, чтобы их столкновение могло привести к химическому взаимодействию.
Рассмотрим путь некоторой элементарной реакции
А + В ––> С
Поскольку химическое взаимодействие частиц связано с разрывом старых химических связей и образованием новых, считается, что всякая элементарная реакция проходит через образование некоторого неустойчивого промежуточного соединения, называемого активированным комплексом:
А ––> K# ––> B
Образование активированного комплекса всегда требует затраты некоторого количества энергии - энергии активации. Она приближенно равна превышению средней энергии активированного комплекса над средним уровнем энергии реагентов. Энергии активации прямой и обратной реакции связаны друг с другом через изменение внутренней энергии в ходе реакции. Вышесказанное можно проиллюстрировать с помощью энергетической диаграммы химической реакции (рис. 2.5).
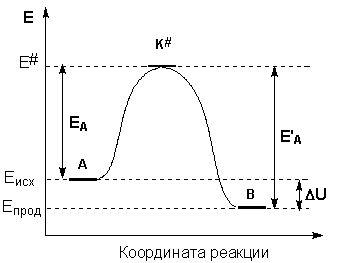
Рис. 2.5. Энергетическая диаграмма химической реакции.
Eисх – средняя энергия частиц исходных веществ,
Eпрод – средняя энергия частиц продуктов реакции
Поскольку температура есть мера средней кинетической энергии частиц, повышение температуры приводит к увеличению доли частиц, энергия которых равна или больше энергии активации, что приводит к увеличению константы скорости реакции.
Зависимость константы скорости реакции от температуры и величины энергии активации определяется уравнением Аррениуса, где EA – энергия активации:
 (II.35)
(II.35)
После интегрирования выражения (II.35) получим уравнение Аррениуса в интегральной форме:
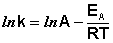 (II.36)
(II.36)
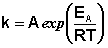 (II.37)
(II.37)
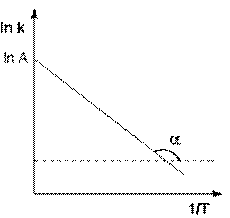
Рис. 2.7 Зависимость логарифма константы скорости химической
реакции от обратной температуры.
Здесь A – постоянная интегрирования. Физический смысл предэкспоненциального множителя A, который равен константе скорости реакции при температуре, стремящейся к бесконечности. Величину энергии активации EA и логарифм предэкспоненциального множителя A можно определить графически (тангенс угла наклона прямой к оси абсцисс и отрезок, отсекаемый прямой на оси ординат).
 (II.38)
(II.38)
Зная энергию активации реакции и константу скорости при какой-либо температуре T1, по уравнению Аррениуса можно рассчитать величину константы скорости при любой температуре T2:
 (II.39)
(II.39)
ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
Передача энергии для активации вступающих во взаимодействие молекул может осуществляться либо в форме теплоты (т. н. темновые реакции), либо в виде квантов электромагнитного излучения. Реакции, в которых активация частиц является результатом их взаимодействия с квантами электромагнитного излучения видимой области спектра, называют фотохимическими реакциями. При всех фотохимических процессах выполняется закон Гротгуса:
Химическое превращение вещества может вызвать только то излучение, которое поглощается этим веществом.
Излучение, отражённое веществом, а также прошедшее сквозь него, не вызывают никаких химических превращений. Иногда фотохимические процессы происходят под действием излучения, которое не поглощается реагирующими веществами; однако в таких случаях реакционная смесь должна содержать т.н. сенсибилизаторы. Механизм действия сенсибилизаторов заключается в том, что они поглощают свет, переходя в возбуждённое состояние, а затем при столкновении с молекулами реагентов передают им избыток своей энергии. Сенсибилизатором фотохимических реакций является, например, хлорофилл.
Между количеством лучистой энергии, поглощенной молекулами вещества, и количеством фотохимически прореагировавших молекул существует соотношение, выражаемое законом фотохимической эквивалентности Штарка – Эйнштейна:
Число молекул, подвергшихся первичному фотохимическому превращению, равно числу поглощенных веществом квантов электромагнитного излучения.
Поскольку фотохимическая реакция, как правило, включает в себя и т.н. вторичные процессы (например, в случае цепного механизма), для описания реакции вводится понятие квантовый выход фотохимической реакции:
Квантовый выход фотохимической реакции γ есть отношение числа частиц, претерпевших превращение, к числу поглощенных веществом квантов света.
Квантовый выход реакции может варьироваться в очень широких пределах: от 10-3 (фотохимическое разложение метилбромида) до 106 (цепная реакция водорода с хлором); в общем случае, чем более долгоживущей является активная частица, тем с большим квантовым выходом протекает фотохимическая реакция.
Важнейшими фотохимическими реакциями являются реакции фотосинтеза, протекающие в растениях с участием хлорофилла.
Процесс фотосинтеза составляют две стадии: световая, связанная с поглощением фотонов, и значительно более медленная темновая, представляющая собой ряд химических превращений, осуществляемых в отсутствие света. Суммарный процесс фотосинтеза заключается в окислении воды до кислорода и восстановлении диоксида углерода до углеводов:
СО2 + Н2О + hν ––> (СН2О) + О2, ΔG° = 477.0 кДж/моль
КАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ
Скорость химической реакции при данной температуре определяется скоростью образования активированного комплекса, которая, в свою очередь, зависит от величины энергии активации. В случае наличия нескольких переходных состояний реакция будет идти в основном по пути с наименьшим активационным барьером.
Катализ – явление изменения скорости химической реакции в присутствии веществ, состояние и количество которых после реакции остаются неизменными.
Различают положительный и отрицательный катализ (соответственно увеличение и уменьшение скорости реакции), хотя часто под термином "катализ" подразумевают только положительный катализ; отрицательный катализ называют ингибированием.
Вещество, входящее в структуру активированного комплекса, но стехиометрически не являющееся реагентом, называется катализатором. Для всех катализаторов характерны такие общие свойства, как специфичность и селективность действия.
Специфичность катализатора заключается в его способности ускорять только одну реакцию или группу однотипных реакций и не влиять на скорость других реакций. Так, например, многие переходные металлы (платина, медь, никель, железо и т.д.) являются катализаторами для процессов гидрирования; оксид алюминия катализирует реакции гидратации и т.д.
Селективность катализатора – способность ускорять одну из возможных при данных условиях параллельных реакций. Благодаря этому можно, применяя различные катализаторы, из одних и тех же исходных веществ получать различные продукты:
| [Cu]: СО + Н2 ––> СН3ОН | [Al2О3]: С2Н5ОН ––> С2Н4 + Н2О |
| [Ni]: СО + Н2 ––> СН4 + Н2О | [Cu]: С2Н5ОН ––> СН3СНО + Н2 |
Причиной увеличения скорости реакции при положительном катализе является уменьшение энергии активации при протекании реакции через активированный комплекс с участием катализатора.
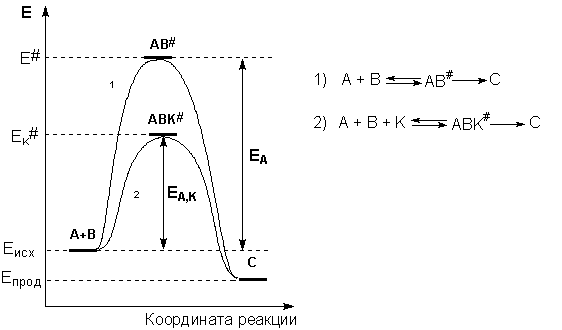
Рис. 2.8. Энергетическая диаграмма химической реакции без катализатора (1) и в присутствии катализатора (2).
Поскольку, согласно уравнению Аррениуса, константа скорости химической реакции находится в экспоненциальной зависимости от величины энергии активации, уменьшение последней вызывает значительное увеличение константы скорости.
Катализатор не изменяет величину константы равновесия для обратимых реакций; влияние катализатора в этом случае заключается только в ускорении достижения равновесного состояния.
В зависимости от фазового состояния реагентов и катализатора различают гомогенный и гетерогенный катализ.
Гомогенный катализ
Гомогенный катализ – каталитические реакции, в которых реагенты и катализатор находятся в одной фазе. В случае гомогенно-каталитических процессов катализатор образует с реагентами промежуточные реакционноспособные продукты. Рассмотрим некоторую реакцию
А + В ––> С
В присутствии катализатора осуществляются две быстро протекающие стадии, в результате которых образуются частицы промежуточного соединения АК и затем (через активированный комплекс АВК#) конечный продукт реакции с регенерацией катализатора:
А + К ––> АК
АК + В ––> С + К
Наиболее распространенным типом гомогенного катализа является кислотный катализ, при котором в роли катализатора выступают ионы водорода Н+.
Автокатализ
Автокатализ – процесс каталитического ускорения химической реакции одним из её продуктов. В качестве примера можно привести катализируемую ионами водорода реакцию гидролиза сложных эфиров. Образующаяся при гидролизе кислота диссоциирует с образованием протонов, которые ускоряют реакцию гидролиза. Особенность автокаталитической реакции состоит в том, что данная реакция протекает с постоянным возрастанием концентрации катализатора. Поэтому в начальный период реакции скорость её возрастает, а на последующих стадиях в результате убыли концентрации реагентов скорость начинает уменьшаться; кинетическая кривая продукта автокаталитической реакции имеет характерный S-образный вид (рис. 2.9).
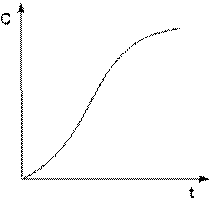
Рис. 2.9. Кинетическая кривая продукта автокаталитической реакции
Гетерогенный катализ
Гетерогенный катализ – каталитические реакции, идущие на поверхности раздела фаз, образуемых катализатором и реагирующими веществами. Механизм гетерогенно-каталитических процессов значительно более сложен, чем в случае гомогенного катализа. В каждой гетерогенно-каталитической реакции можно выделить как минимум шесть стадий:
1. Диффузия исходных веществ к поверхности катализатора.
2. Адсорбция исходных веществ на поверхности с образованием некоторого промежуточного соединения:
А + В + К ––> АВК
3. Активация адсорбированного состояния (необходимая для этого энергия есть истинная энергия активации процесса):
АВК ––> АВК#
4. Распад активированного комплекса с образованием адсорбированных продуктов реакции:
АВК# ––> СDК
5. Десорбция продуктов реакции с поверхности катализатора.
СDК ––> С + D + К
6. Диффузия продуктов реакции от поверхности катализатора.
Специфической особенностью гетерокаталитических процессов является способность катализатора к промотированию и отравлению.
Промотирование – увеличение активности катализатора в присутствии веществ, которые сами не являются катализаторами данного процесса (промоторов). Например, для катализируемой металлическим никелем реакции
СО + Н2 ––> СН4 + Н2О
введение в никелевый катализатор небольшой примеси церия приводит к резкому возрастанию активности катализатора.
Отравление – резкое снижение активности катализатора в присутствии некоторых веществ (т. н. каталитических ядов).
Для объяснения этих особенностей гетерогенно-каталитических процессов Г. Тэйлором было высказано следующее предположение: каталитически активной является не вся поверхность катализатора, а лишь некоторые её участки – т.н. активные центры, которыми могут являться различные дефекты кристаллической структуры катализатора (например, выступы либо впадины на поверхности катализатора). В настоящее время нет единой теории гетерогенного катализа. Для металлических катализаторов была разработана теория мультиплетов. Основные положения мультиплетной теории состоят в следующем:
1. Активный центр катализатора представляет собой совокупность определенного числа адсорбционных центров, расположенных на поверхности катализатора в геометрическом соответствии со строением молекулы, претерпевающей превращение.
2. При адсорбции реагирующих молекул на активном центре образуется мультиплетный комплекс, в результате чего происходит перераспределение связей, приводящее к образованию продуктов реакции.
Теорию мультиплетов называют иногда теорией геометрического подобия активного центра и реагирующих молекул. Для различных реакций число адсорбционных центров (каждый из которых отождествляется с атомом металла) в активном центре различно – 2, 3, 4 и т.д. Подобные активные центры называются соответственно дублет, триплет, квадруплет и т.д. (в общем случае мультиплет, чему и обязана теория своим названием).
Ферментативный катализ.
Ферментативный катализ – каталитические реакции, протекающие с участием ферментов – биологических катализаторов белковой природы. Ферментативный катализ имеет две характерные особенности:
1. Высокая активность, на несколько порядков превышающая активность неорганических катализаторов, что объясняется очень значительным снижением энергии активации процесса ферментами.
2. Высокая специфичность. Например, амилаза катализирует процесс расщепления крахмала, представляющего собой цепь одинаковых глюкозных звеньев, но не катализирует гидролиз сахарозы, молекула которой составлена из глюкозного и фруктозного фрагментов.
Согласно о
|
|
|
