 |


Окисление ЖК в пероксисомах
|
|
|
|
В пероксисомах β-окисления ЖК протекает в модифицированной форме. Этот путь обеспечивает катаболизм в печени длинноцепочечных ЖК (С=20, 22) и не сопряжен с образованием АТФ. Продуктами окисления является актоноил-КоА, Ацетил-КоА и Н2О2. Н2О2 синтезируется аэробной дегидрогеназой при взаимодействии ФАДН2 и О2. Актоноил и Ацетил переходят с КоА на карнитин и направляются в митохондрии, где окисляются.
α-окисление ЖК
α-окисление — специфический путь катаболизма ЖК с длинной (более 20 атомов С) и разветвленной углеводородной цепью. α-окисления протекает в нервной ткани, где преобладают ЖК с длинной цепью и в печени, куда поступают разветвленные ЖК растительной пищи (например, фитановая кислота).
При α-окислении синтез АТФ не происходит, от ЖК отщепляется по одному атому С, в виде СО2.

Фитановая кислота, ЖК с разветвлённой углеводородной цепью, образуется из фитола, который входит в состав хлорофилла. В этой кислоте у каждого третьего атома С находится метильная группа, что делает невозможным β-окисление данной кислоты. При α-окислении фитановой кислоты вначале удаляется метильная группа, а затем происходит цикл β-окисления.
ω-Окисление ЖК
ω-Окисление протекает в ЭПР, начинается с гидроксилирования ω-углеродного атома ЖК монооксигеназой (Р450) и в результате окисления приводит к образованию ЖК с двумя карбоксильными группами, которые разрушаются β-окислением с обеих сторон до дикарбоновых кислот: адипиновой (С6) и субериновой кислоты (С8), которые, выводятся с мочой.
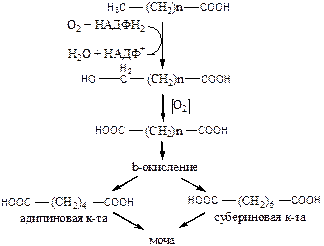
Нарушения окисления ЖК
1) Нарушение β-окисления возникает при снижении транспорта ЖК в митохондрии.
Скорость переноса ЖК внутрь митохондрий зависит от доступности карнитина и активности карнитинацилтрансферазы I. Снижение карнитина происходит при длительном гемодиализе, длительной ацидурии (карнитин выводится как основание с органическими кислотами), нарушении синтеза карнитина. Карнитинацилтрансфераза I ингибируется препаратами сульфонилмочевины (лечение больных сахарным диабетом), существуют наследственные дефекты карнитинацилтрансферазы I.
|
|
|
В этих случаях ЖК с длинной цепью не используются как источники энергии. У таких людей снижена способность к физической активности; в мышечных клетках могут накапливаться ТГ в виде липидных капель.
2) Генетический дефект дегидрогеназы ЖК со средней длиной углеводородной цепи.
В митохондриях имеется 3 вида ацил-КоА-дегидрогеназ, окисляющих ЖК с длинной, средней или короткой цепью радикала. ЖК по мере укорочения радикала в процессе β-окисления последовательно окисляются этими ферментами.
Генетический дефект дегидрогеназы ЖК со средней длиной радикала - распространенное аутосомно-рецессивное заболевание (1:15 000). Частота дефектного гена в европейской популяции — 1:40.
Активность этой дегидрогеназы особенно важна для грудных детей, т.к. у них основным источником энергии служат ЖК со средней длиной цепи, входящие в состав ТГ молока.
Невозможность использовать ЖК как источники энергии приводит к увеличению скорости окисления глюкозы. В результате у детей развивается гипогликемия, которая является причиной внезапной смерти (10% от общего числа умерших новорождённых). Если такие дети выживают, то после голодания в течение 6—8 ч у них развиваются гипогликемические приступы (слабость, головокружение, рвота, потеря сознания). Введение глюкозы приводит к исчезновению симптомов.
Во всех случаях, когда нарушается β-окисление, ЖК накапливаются в клетках и распадаются по пути ω-окисления, которое в норме идёт с очень низкой скоростью. Окисление происходит по метильному сжатому углерода, и в результате образуются дикарбоновые кислоты, выделяющиеся с мочой. Определение этих кислот в моче может служить диагностическим признаком нарушения β-окисления.
|
|
|
3) Нарушение α-окисления
Болезнь Рефсума - редкое наследственное заболевание развивающейся вследствие генетических дефектов ферментов α-окисления. Фитановая кислота, поступающая с пищей, не окисляется и накапливается в организме, в основном в нервной ткани.
Это приводит у взрослых к нарушению структуры нервной ткани и развитию многих неврологических симптомов (полиневропатии, мозжечковой атаксии), нарушению зрения (пигментная дистрофия сетчатки), костным деформациям, изменениям кожи по типу ихтиоза и поражениям сердца с развитием кардиомиопатий.
У детей развиваются выраженные черепно-лицевые аномалии, мышечная гипотония, пигментный ретинит, нейросенсорная глухота, грубая задержка психомоторного развития, судороги, гепатомегалия с нарушением функции печени.
4) Нарушение деградации ЖК в пероксисомах
Из-за дефекта пероксисом нарушена деградация длинноцепочечных жирных кислот, что проявляется в синдроме Целльвегера. В клетках отмечается повышение количества жирных кислот с длинной углеводной цепью.
Синдром Целльвегера - заболевание раннего детского возраста, наследующееся по аутосомно-рецессивному типу и проявляющееся мышечной гипотонией, нарушением моторики, арефлексией, кардиомиопатией, задержкой психического развития, судорогами, фиброзом печени и кистозом почек. Характерны черепно-лицевые дизморфии, атрофии зрительных нервов, помутнение хрусталика и роговицы, глаукома. С первых месяцев жизни выявляется выраженная задержка психомоторного развития.
|
|
|
