 |


При сумме более 5 баллов случай подозрителен на врожденный гипотиреоз.
|
|
|
|
Если диагноз гипотиреоза не был установлен своевременно, то рост и развитие ребенка задерживаются. Задержка роста заметна уже в возрасте 3—4 мес. Следует ожидать также задержки психического развития и появления неврологических симптомов - атаксии, нарушения координации движений, пирамидных симптомов (спастическая диплегия), гипотонии и спастичности мышц, нейросенсорной тугоухости и косоглазия. Наблюдаются сниженный аппетит, затруднения при глотании, плохая прибавка в массе тела; метеоризм, запоры; сухость, бледность, шелушение кожных покровов; гипотермия (холодные кисти, стопы); сухие, тусклые волосы, мышечная гипотония.
В более поздние сроки, после 5-6 месяцев жизни на первый план выступает нарастающая задержка психомоторного и физического развития. Пропорции тела у детей с ВГ приближаются к хондродистрофическим, отстает развитие лицевого скелета (широкая запавшая переносица, гипертелоризм, позднее закрытие родничков). Запаздывает прорезывание, а позднее и смена зубов. У больных ВГ отмечается кардиомегалия, глухость сердечных тонов, снижение артериального давления, брадикардия.
При рентгенологическом исследовании конечностей отмечается задержка появления ядер окостенения, их ассиметрия, нарушение последовательности появления. Патогномоничным признаком является эпифизарный дисгенез.
Страдает умственное развитие: ребенок поздно и с трудом узнает мать, с большим опозданием развивается речь. При отсутствии адекватного лечения возникает нарушение интеллекта, которое носит необратимый характер, иногда вплоть до развития олигофрении.
Заподозрить гипотиреоз у новорожденного только на основании клинических признаков удается не более чем в 5-10% случаев.
|
|
|
Диагностика ВГ основывается на данных гормонального статуса (гипертиреотропинемия и гипотироксинемия), результатах УЗИ щитовидной железы (аплазия, гипоплазия, дистопия) и радиоизотопного сканирования. При отсутствии визуализации щитовидной железы при проведении радиоизотопного сканирования диагноз не вызывает сомнений. Этот метод исследования (в отличие от УЗИ) позволяет выявить дистопически расположенную ткань щитовидной железы. В диагностических целях высокоинформативным методом является определение тиреоглобулина в сыворотке крови как маркера наличия ткани щитовидной железы
Неонатальный скрининг. Протокол обследования на ВГ включает:
1) первичный тест - при выявлении концентрации ТТГ в сухом пятне крови выше 20 мкМЕ/мл проводится повторное обследование;
2) ретест - если при повторном анализе уровень ТТГ в крови превышает 20 мкМЕ/мл, то новорожденному устанавливается диагноз врожденный гипотиреоз.
Лечение. Принимая во внимание высокую чувствительность фетального и неонатального мозга к недостаточности тиреоидных гормонов, проведение заместительной гормональной терапии является неотложным мероприятием и осуществляется препаратами левотироксина, с учетом индивидуальных клинических и лабораторных данных.
К настоящему времени установлено несколько факторов, влияющих на IQ у детей с ВГ. Наиболее значимыми из них оказались доза L-T4 и время начала лечения (таблица 6). Однако, появились данные, свидетельствующие о том, что прогноз интеллектуального развития ребенка с ВГ зависит также от других причин: тяжесть пренатального дефицита тиреоидных гормонов, адекватность проводимой заместительной гормональной терапии (особенно в первые годы жизни ребенка), патология беременности и родов, наследственность и социальное окружение.
Таблица 6. Дозы L-тироксина для лечения детей с ВГ
|
|
|
| Возраст | L-тироксин, мкг/сут | L-тироксин, мкг/кг/сут |
| Для недоношенных | 8-10 | |
| 0-3 месяца | 15-50 | 10-15 |
| 3-6 месяцев | 25-50 | 8-10 |
| 6-12 месяцев | 50-75 | 6-8 |
| 1-3 года | 75-100 | 4-8 |
| 3-10 лет | 100-150 | 4-6 |
| 10-15 лет | 100-150 | 3-4 |
| Старше 15 лет | 100-200 | 2-3 |
В комплекс лечебных мероприятий при ВГ следует включать симптоматическую терапию (антианемическая, антирахитическая, витаминотерапия), ЛФК, массаж, ноотропные препараты.
Прогноз для жизни, интеллекта и репродукциипри условии своевременно начатого и адекватного лечения - благоприятный.
Врожденная гиперплазия коры надпочечников (ВГКН) (адреногенитальный синдром, врожденная дисфункция коры надпочечников) - группа заболеваний, в основе которых лежит дефект одного из ферментов или транспортных белков, принимающих участие в биосинтезе кортизола в коре надпочечников. Хронический дефицит кортизола приводит к повышению секреции АКТГ, развитию гиперплазии коры надпочечников и накоплению метаболитов, предшествующих дефектному этапу стероидогенеза, а также к активации альтернативных путей синтеза стероидов.
Классификация. Выделяют шесть клинических форм ВДКН, в основе классификации лежат нарушения ферментов катализирующих различные этапы стероидогенеза, обусловленные дефектами семи генов:
1. Липоидная гиперпалазия надпочечников (дефект StAR-протеина или синдром Прадера) – гены SТAR и CYP11А;
2. Дефицит 3βГСД (3β-гидроксистероиддегидрогеназы) – ген HSD3B2;
3. Дефицит Р450с17 (17α-гидроксилазы/17,20-лиаза или синдром Беглиери) – ген CYP17А1;
4. Дефицит Р450с21 (21-гидроксилазы)– ген CYP21А2;
5. Дефицит Р450с11 (11β -гидроксилазы) – ген CYP11В1;
6. Дефицит Р450 оксидоредуктазы – ген РОR.
Наиболее распространенными являются формы обусловленные дефектами генов CYP21А2 (более 90%) и CYP11В1 (около 5%), контролирующие надпочечниковый стероидогенез. Мутации генов HSD3B2,CYP17А1, CYP17А1, SТAR и РОR встречаются гораздо реже и влекут за собой нарушение стероидогенеза надпочечниках и в гонадах.
Три формы ВДКН приводят к развитию первичной надпочечниковой недостаточности с минералкортикоидным компонентом: липоидная гиперпалазия надпочечников, дефицит 3βГСД, дефицит CYP21.
Дефицит 21-гидроксилазы (Р450с21) -одно из самых частых врожденных ферментативных нарушений стероидогенеза, составляет более 90-95% всех случаев ВДКН. Клинически известны два классических варианта недостаточности 21-гидроксилазы (сольтеряющая и простая вирильная форма) и один неклассический.
|
|
|
Популяционная частота классических вариантов заболевания, рассчитанная по данным неонатального скрининга, в различных популяциях колеблется от 1:10000 до 1:18000 новорожденных. Чрезвычайно высокая частота выявлена в двух изолированных популяциях: у эскимосов Юппи, проживающих в западной Аляске - 1:280 и у жителей острова Ла Руньон в Индийском океане — 1:2100.
Частота встречаемости неклассической формы дефицита 21-гидроксилазы в общей популяции очень высока до - 0,3%, в среднем колеблется от 1:27 до 1:333. А в некоторых этнических группах еще выше: 1,6% в Югославии, 1,9% в Испании, 3,7% у евреев западной Европы (Ashkenazi).
Этиология. Ген 21-гидроксилазы (CYP21A2) картирован на коротком плече хромосомы 6 (6р21.3) внутри сложного комплекса HLA-генов, кодирующих белки главного комплекса гистосовместимости. В настоящее время известно около 100 мутаций, обусловливающих развитие ВДКН.
Для пренатальной диагностики молекулярно-генетический метод является единственным достоверным способом выявления заболевания и его формы у плода.
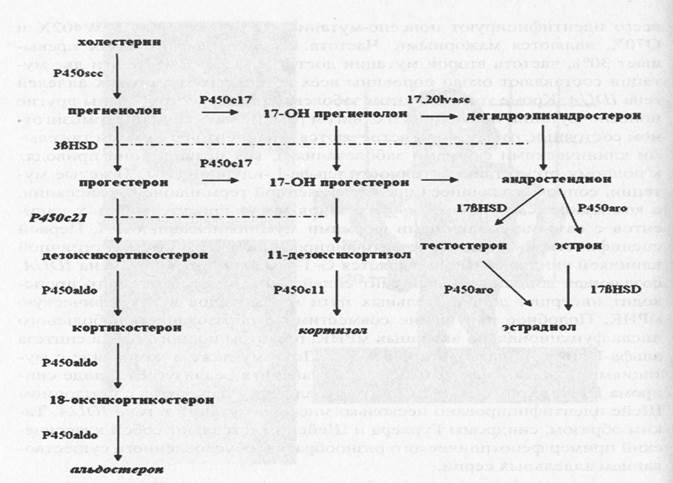
Патогенез. При недостаточности 21-гироксилазы блокируется превращение 17-гидроксипрогестерона в 11-дезоксикортизол и прогестерона в дезоксикортикостерон. В первом случае происходит снижение образования кортизола и накопление его предшественников, таких, как 17-гидроксипрогестерон и 17-гидроксипрегненолон. Во втором — возникает дефицит альдостерона и накапливаются прегненолон и прогестерон. Их избыток в сетчатом слое коры надпочечников приводит к повышению уровня надпочечниковых андрогенов. Дефицит 21-гидроксилазы приводит к недостаточной продукции кортизола, что вызывает повышение секреции АКТГ и приводит к гиперплазии надпочечников (рис. 3).
Минералокортикоидная недостаточность различной степени выраженности выявляется удетей с дефицитом 21-гидроксилазы. Снижение уровня дезоксикортикостерона и альдостерона приводит к снижению реабсорбции натрия в почках, концентрация натрия в сыворотке крови падает и возрастает почечная реабсорбция калия. Результатом этих нарушений является гипонатриемия, гиперкалиемия, ацидоз, потеря жидкости. В ответ на снижение выработки минералокортикоидов возрастает рениновая активность плазмы, повышается уровень ангиотензина II.
|
|
|
|
Рисунок 3. Метаболизм стероидов и генетические варианты адреногенитального синдрома.
|
|
|
