 |


Кетоновые тела. Приобретенная. Врожденная
|
|
|
|
Кетоновые тела
Проявления нарушений обмена липидов очень сильно выражены при сахарном диабете. Основными нарушениями обмена липидов при диабете являются усиление катаболизма липидов, увеличение образование кетоновых тел, снижение синтеза жирных кислот и триацилглицеридов. Кровеносное русло заполняется триацилглицеролами и свободными жирными кислотами. Так как ингибируется синтез жирных кислот, катаболические процессы не могут справиться с избытком образующегося ацетил-коА. В печени ацетил-коА служит источником синтеза кетоновых тел. Кетоновые тела накапливаются в крови в связи с тем, что скорость их образования превышает возможности клеток по их использованию.
Избыток ацетил-коА обычно используется в синтезе ацетоацетил-коА, который в печени превращается в ацетоацетат и затем в ацетон или в-гидроксибутират.
(См. главу К. Г. М)
Образующиеся в больших количествах кетоновые тела поступают в кровообращение. Циркулирующие кетоновые тела - важный источник энергии при голодании. Около половины энергетических затрат при голодании обеспечивается окислением кетоновых тел. При сахарном диабете доля используемых в качестве источника энергии кетоновых тел также значительна.
|
Приобретенная |
Врожденная
| ||||
| Патология обмена нейтральных жиров: | Патология обмена холестерола | Дислипопро- теинемия | Сфинголипи- дозы | ||
| Ожирение | Желчекаменная болезнь | Болезнь Гоше | |||
| Жировое перерождение печени |
Атеросклероз
| Болезнь Ниманна-Пика | |||
|
| Болезнь Тея-Сакса | ||||
| Болезнь Фабри | |||||
| Болезнь Краббе | |||||
Липидозы.
Существует не менее десятка наследственных сфинголипидозов, нарушающих раннее психомоторное развитие детей. Сфинголипидозы это наследственные заболевания, при которых нарушен катаболизм сфингомиелинов. Эти заболевания передаются по аутосомно-рецессивному типу. Это значит, что заболевание может возникнуть у того ребенка который унаследовал одновременно два дефектных гена, один ген то матери, а второй от отца. Если присутствует дефектный ген у одного из родителей, то ребенок не заболеет, а с 50% вероятностью становится носителем дефектного гена. Если дефектный ген есть у одного из родителей, малыш будет здоров, но с 50% вероятностью будет носителем, что ставит в будущем под угрозу здоровье его наследников. При наличии гена с дефектом у обоих родителей возможно три варианта развития событий. Ребёнок с вероятность 25 % может появиться на свет здоровым и не будет носителем гена. В 50% случаев малыш будет носителем дефектного гена, но родится здоровым. В 25% случаев малышу может достаться два гена с дефектом, и он родиться больным.
Следует отметить, что эти наследственные болезни нарушения липидного обмена характерны для определенных этнических групп.
Механизм развития сфинголипидозов заключается в медленном накапливании липидов в нервной системе. В здоровом организме эти сфинголипиды постоянно синтезируются и расщепляются. За нормальное поддержание равновесия между распадом и синтезом сфинголипидов отвечают их ферментные системы. Больной организм, который имеет поврежденный ген, не может синтезировать тот фермент, который отвечает за распад сфинголипидов. Вследствие чего происходит постепенное накопление сфинголипидов, которые быстро откладываются в мозгу, что блокирует работу нервных клеток, и это приводит к самым тяжелым последствиям. Наиболее распространенными формами сфинголипидозов являются болезни Гоше, Ниманна-Пика, Тея-Сакса.
|
|
|
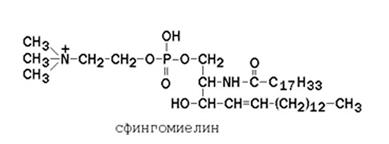
Сфинголипиды - основные структурные компоненты клеточных мембран, особенно богаты сфинголипидами миелин. Основу сфинголипидов составляет сфингозин. Сфингозин образуется в результате взаимодействия пальмитоил-коА и серина. При N-ацетилировании сфингозина образуются церамиды, которые входят в состав многих сфинголипидов, например сфингомиелина. Кроме того церамид содержится в цереброзидах и ганглиозидах. Цереброзиды и ганглиозиды содержат остатки углеводов. Сфингомиелин состоит из, церамида, холина и фосфорного остатка.
Цереброзиды образуется при связывании моносахарида с церамидом. Например, при связывании глюкозы с церамидом образуется глюкоцереброзид, а при связывании галактозы – галактозилцереброзид. Глобозиды – цереброзиды содержащие несколько углеводных остатков.
Болезнь Гоше. Заболевание наследуется по аутосомно-рецессивному типу.
Причина заболевания – дефект гена, ответственного за синтез фермента β - глюкоцереброзидазы. Дефект и дефицит этого фермента ведут к нарушению утилизации глюкоцереброзидов и накоплению их в головном мозге, селезенке, печени, костном мозге.
Диагноз устанавливают по обнаружению в пунктате селезенки или костном мозге, так называемых, клеток Гоше. Эти клетки имеют лимфоцитоподобное ядро с очень широкой и светлой цитоплазмой с циркулярной исчерченностью. Наиболее часто болезнь Гоше встречается у евреев западноевропейской группы Ашкенази.
Выделяют три типа болезни Гоше. Существование нескольких разнообразных форм болезни обьясняется гетерогенностью мутаций гена – β - гликозидазы.
Тип 1. При первом типе неврологические нарушения отсутствуют. Головной и спинной мозг не поражаются. Симптомы: умеренное увеличение печени и селезенки, анемия, тромбоцитопения и лейкопения, поражение почек. Повреждение костной ткани: частые переломы, деформация бедренной кости. Низкий рост. Больные с этим типом заболевания могут прожить достаточно долго. Возможно «бессимптомное» течение.
|
|
|
Тип 2. Злокачественная форма. Обширные повреждения головного мозга: судороги и повышение мышечного тонуса конечностей, задержка умственного и физического развития, нарушения движения глаз, нарушения процессов глотания и дыхания. Увеличение печени и селезенки. Возможна спонтанная остановка дыхания. Обычно умирают в возрасте от одного до двух лет.
Тип 3. Менее злокачествен, чем тип 2. Первые симптомы появляются в возрасте от 6 до 15 лет. Неврологические симптомы: судороги, нарушение глотания, непроизвольные сокращения и спазмы мышц лица. Развивается косоглазие и затрудняется дыхания на вдохе. Длительность жизни больных детей до 17 л. (редко доживают до 30-40 лет). Характерно прогрессирование заболевания. Смерть наступает в результате осложнений пораженных органов: печени и легких.
В настоящее время лечение 1 и 3 типа данной болезни осуществляется на основе ферментотерапии, которая заключается систематическом внутривенном введении рекомбинантной β -глюкоцереброзидазы. Также в лечении используют препараты – имиглюцеразу, velaglucerase alfa, miglustat, taliglucerase alfa. Для облегчения симптомов болезни применяют обезболивающие, противосудорожные препараты.
Возможно использование генной терапии основанной на переносе гена β - глюкоцереброзидазы в ДНК кроветворных стволовых клеток.
Болезнь Краббе названа в честь датского невролога Краббе, который описал её 1916 г.
Причина развития. Болезнь вызвана мутациями в GALC в гене, который расположен на 14 хромосоме в регионе g31. Ген GALC обеспечивает синтез фермента галактоцерамидазы (галактозилцерамид-в-галактозидазы). Этот фермент расщепляет гликолипид галактоцереброзид на галактозу и церамид.
Заболевание проявляется в поражении миелиновой оболочки нервных волокон, повышении мышечного тонуса, высокой температуре тела (гиперпирексии) и умственной отсталости.
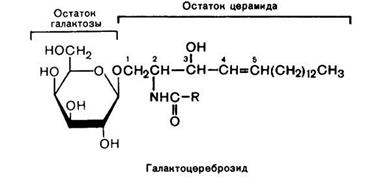
Рис. 20 Строение галактоцереброзида.
Галактоцереброзид является важным компонентом миелина, который образует защитное покрытие вокруг нервных волокон, обеспечивающих быструю передачу нервных импульсов. Вследствие происходит накопление большого количества нерасщепленной производной галактоцереброзида психозина – галактозилсфингозин. Накопление психозина токсично для клеток, формирующих миелиновую оболочку, поэтому она постепенно разрушается. В результате процесс дегенерации затрагивает не только ЦНС, но и периферические нервы. Для нервной системы психозин токсичен, поскольку он вызывает гибель клеток нейроглии – олигодендроцитов, которые обеспечивают миелинизацию аксонов. В зонах распада миелиновой оболочки в нервной ткани вокруг кровеносных сосудов образуются характерные включения – глобоидные гистиоциты (являются макрофагами, которые способны захватывать и переваривать бактерии и т. д. ).
|
|
|
Гибель олигодендроцитов сопровождается повреждением нейронов, которые являются основной структурно-функциональной единицей мозга. Место отмерших нейронов заполняется клетками нейроглии и развивается глиоз.
Болезнь Фабри (синдром Андерсона – Фабри) описан врачами дерматологами в 1898 году.
Болезнь Фабри является Х-сцепленной.
Причина заболевания – структурная мутация гена, расположенного на Х-хромосоме, который отвечает за синтез фермента α -галактозидазы А. Этот фермент а-1, 4 галактозилгидролаза, отщепляет концевой галактозильный остаток в тригексозилцерамиде. В случае отсутствия этого фермента происходит накопление церамидтригексозида ( глоботриаозилцерамида - Gb3 ) в стенках кровеносных сосудов, сердечной мышце, почечных канальцах и клубочках, нервной системе, скелетной мускулатуре. Такое накопление приводит к нарушению их нормального функционирования.
Симптомы: боли и парестезия конечностей; кожные изменения: макулопапулезные высыпания на ягодицах, в области пупка, паховой области, в области губ и пальцев рук.
Поражения со стороны:
1. глаз: эктазия конъюнктивальных сосудов, отек век, помутнение роговой оболочки;
2. почек: гематурия, протеинурия, цилиндрурия;
3. сердца: кардиомегалия, инфаркт миокарда, артериальная гипертония;
4. пищеварительной системы: боли в животе, диарея;
5. нервной системы: мозговые кровоизлияния с частично выраженным параличом.
Боль в желудочно-кишечном тракте, вероятно, обусловлена накоплением липидов в его малых сосудах, что мешает кровообращению (ишемия кишечника), вызывая боль. Сильные боли в конечностях связаны с повреждением периферических нервных волокон, передающих боль.
|
|
|
Боли при болезни Фабри могут носить характер «кризов», в виде приступов. Для пациентов с болезнью Фабри характерно отсутствие потоотделения (ангидроз).
Болезнь Ниманна – Пика. Наследование болезни происходит по аутосомно-рецессивному типу.
Причина заболевания структурная мутация гена SMPD1, который отвечает за синтезфермента сфингомиелиназы. В случае отсутствия фермента происходит накопление сфингомиелина в ретикуло-эндотелиальных клетках различных органов и тканей. Болезнь Нимана - Пика характеризуется тяжелым поражением нервной системы, задержкой общего развития ребенка.
Микроскопически в печени, селезенке, костном мозге, почках, надпочечниках, лимфатических узлах, и некоторых других органах обнаруживаются «пенистые» клетки Нимана-Пика (они так выглядят из-за накопления жиров). Это крупные клетки, размером от 20 до 50 мк и содержащие одно или много ядер. Протоплазма клеток содержит вакуоли, придающие клеткам пенистый вид.
Клинически болезнь Нимана — Пика проявляется обычно в грудном возрасте, чаще в первое полугодие. Начальными симптомами болезни являются потеря аппетита, резкое похудание ребенка, задержка физического и умственного развития. Живот больного значительно увеличивается вследствие гепатоспленомегалии (увеличение селезенки и печени). Часто возникают бронхопневмонии. Лимфатические железы в большинстве случаев увеличиваются. Кожные покровы выглядят восковидными, блестящими, обнаруживаются участки пигментации. В начальных стадиях болезни выявляются двигательные расстройства: парезы конечностей, повышение тонуса мышц и сухожильных рефлексов, пирамидные знаки. В более поздних стадиях характерна гипотония мышц, отсутствие сухожильных рефлексов. Развивается идиотия, слепота и глухота. На глазном дне у многих больных могут обнаруживаться атрофия сосков зрительного нерва, вишнево-красное пятнышко овальной формы в макулярной области.
Болезнь Нимана — Пика обладает злокачественным течением. Большинство детей погибают в первые 2 года жизни от легочно-сердечной недостаточности, интеркуррентных инфекций.
На сегодня заболевание классифицируется следующим образом:
Тип А – самый тяжёлый тип. Проявляется в грудном возрасте. Характеризуется увеличением печени и селезёнки (гепатоспленомегалия) и прогрессивным поражением нервной системы: повышение тонуса мышц и сухожильных рефлексов, сильные боли и паралич конечностей. Начальные симптомы: потеря аппетита, резкое похудание ребенка, задержка психофизического развития. Дети погибают в первые 2 года жизни.
Тип B – умеренный тип. Характерны гепатоспленомегалия, тромбоцитопения, задержка роста и нарушение лёгочной функции с частыми лёгочными инфекциями.
Тип С – проявляется и в детстве и возможно у взрослых. Симптомы: тяжёлые печёночные нарушения, проблемы с дыханием, задержка в развитии, припадки, повышенный мышечный тонус (дистония), нарушение координации движения, нарушения питания и характерны движения глаз в вертикальной плоскости.
Этот тип С вызывается мутациями генов NPC1 или NPC2, которые кодируют белок клеточной мембраны, отвечающий за транспорт холестерина и липидов внутри клетки.
Заболевание неизлечимо. В основном проводится симптоматическое лечение для облегчения страданий больного.
Болезнь Тея-Сакса – наследственное заболевание, связанное с накоплением в головном мозге ганглиозида GM2.
Болезнь Тея-Сакса обусловлена мутационными поражениями гена гексозаминидазы (HEXA), контролирующего синтез альфа субъединицы гексозаминидазы А. Гексозаминидаза А - лизосомный фермент, катализирующий катаболизм GM2 ганглиозида.
Различают три формы болезни Тея — Сакса:
Детская форма – через полгода после рождения у детей отмечается ухудшение физических возможностей и умственных способностей. Наблюдаются слепота, глухота, потеря способности глотать. В результате атрофии мышц развивается паралич. Смерть наступает в возрасте до 3—4 лет.
Подростковая форма характеризуется развитием моторно-когнитивных проблем, нарушением глотания, расстройством речи, параличом конечностей. Смерть наступает в возрасте до 15–16 лет.
Взрослая форма – возникает в возрасте от 25 до 30 лет. Характеризуется симптомами прогрессирующего ухудшения неврологических функций: нарушение и шаткость походки, расстройства глотания и речи, снижение когнитивных навыков.
.
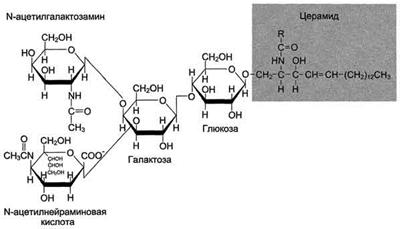
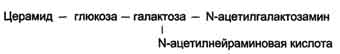
Рис. № 21. Строение ганглиозида GM2
Таблица «Наследственные нарушения липидного обмена» (см. отд. док. №1)
Синтез и распад кетоновых тел.
В условиях голодания, при длительной физической нагрузки и нарушения утилизации глюкозы клетками (вследствие дефицита инсулина при сахарном диабете) основным источником энергии становятся жирные кислоты, которые образуются при гидролизе триглицеридов жировой ткани. Жирные кислоты подвергаются b-окислению с образованием больших количеств ацетил-КоА во всех органах, кроме мозга. Оксалоацетата, необходимого для включения ацетил-КоА в цикл Кребса не достаточно, поэтому избыток ацетил-КоА не может быстро окислиться в ЦТК. Печень обладает способностью синтезировать из больших количеств ацетил-КоА кетоновые тела.
К кетоновым телам относят три соединения близкой структуры – ацетоацетат, 3-гидроксибутират (b-гидроксибутират) и ацетон:
|
|
|
