врожденные пороки сердца со сбросом крови слева направо.
III. Нарушение наполнения желудочков сердца.
1.Стеноз левого или правого атриовентрикулярного отверстия.
2.Экссудативный и констриктивный перикардит.
3.Перикардиальный выпот (тампонада сердца).
4.Заболевания с повышенной жесткостью миокарда и диастолической дисфункцией:
гипертрофическая кардиомиопатия (ГКМП);
амилоидоз сердца;
фиброэластоз;
эндомиокардиальный фиброз;
выраженная гипертрофия миокарда, в том числе при аортальном стенозе, АГ и других заболеваниях.
IV. Повышение метаболических потребностей тканей (СН с высоким МО).
1.Гипоксические состояния:
анемии;
хроническое легочное сердце.
2.Повышение обмена веществ:
гипертиреоз.
3.Беременность.
Запомните
Наиболее частыми причинами сердечной недостаточности являются: ИБС, включая острый ИМ и постинфарктный кардиосклероз; артериальная гипертензия, в том числе в сочетании с ИБС; клапанные пороки сердца.
2.2. Патогенез
2.2.1. Механизмы систолической и диастолической дисфункции желудочков
Многообразие причин сердечной недостаточности объясняет существование различных клинических и патофизиологических форм этого патологического синдрома, при каждой из которых преобладает преимущественное поражение тех или иных отделов сердца и действие различных механизмов компенсации и декомпенсации. В большинстве случаев (около 70–75%) речь идет о преимущественном нарушении систолической функции сердца, которая определяется степенью укорочения сердечной мышцы и величиной сердечного выброса (МО).
Напомним, что УО и МО определяются тремя гемодинамическими факторами.
1. Исходной длиной мышечного волокна, или конечно-диастолическим объемом (КДО) желудочка, т.е. величиной преднагрузки, которая, в свою очередь, зависит от объема циркулирующей крови (ОЦК), притока крови к сердцу, эффективности сокращения предсердий и других факторов.
2.Инотропным состоянием (сократимостью) миокарда желудочков, которое зависит от активности САС, частоты сердечных сокращений (ЧСС), массы функционирующего миокарда, состояния обменных процессов в кардиомиоцитах, величины коронарной перфузии и т.д.
3. Внутримиокардиальным напряжением, которое должна развивать сердечная мышца во время своего сокращения, чтобы преодолеть сопротивление изгнанию крови, т.е. величиной постнагрузки. Напряжение миокарда, в свою очередь, зависит от уровня давления в аорте или легочной артерии, массы функционирующего миокарда, от размеров полости желудочка и т.п.
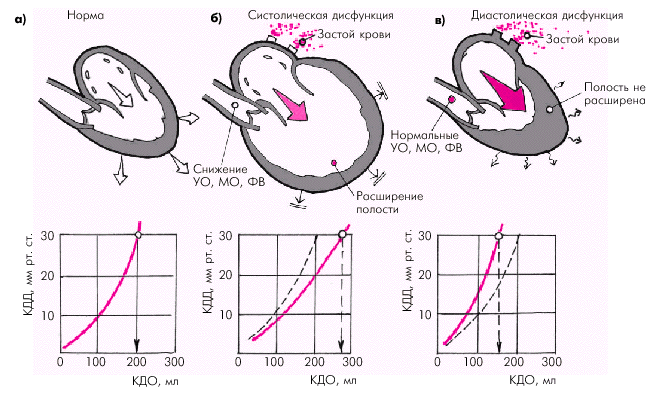
На конечных этапах развития систолической дисфункции наиболее характерную последовательность гемодинамических изменений можно представить следующим образом (рис. 2.1, б):
снижение УО, МО и ФВ, что сопровождается возрастанием конечно-систолического объема (КСО) желудочка, а также гипоперфузией периферических органов и тканей;
возрастание конечно-диастолического давления (КДД) в желудочке, т.е. давления наполнения желудочка;
миогенная дилатация желудочка — увеличение конечно-диастолического объема (КДО) желудочка;
застой крови в венозном русле малого или большого круга кровообращения.
Последний гемодинамический признак СН сопровождается наиболее “яркими” и четко очерченными клиническими проявлениями СН (одышка, отеки, гепатомегалия и т.п.) и определяет клиническую картину двух ее форм. При левожелудочковой СН развивается застой крови в малом круге кровообращения, а при правожелудочковой СН — в венозном русле большого круга.
Быстрое развитие систолической дисфункции желудочка приводит к возникновению острой СН (лево- или правожелудочковой). Такая ситуация возникает, например, при остром повреждении сердечной мышцы (ИМ, миокардит и др.) или внезапном возрастании величины преднагрузки (разрыв межжелудочковой перегородки или папиллярной мышцы при ИМ, введение в сосудистое русло больших количеств жидкости) или постнагрузки (резкий подъем АД при гипертоническом кризе или тромбоэмболия легочной артерии, сопровождающаяся возрастанием давления в ЛА).
Рис. 2.1. Особенности функции ЛЖ при систолической (б) и диастолической (в) формах ХСН. КДД ЛЖ, равное 30 мм рт. ст., — критическое давление наполнения, при котором развивается отек легких.
Внизу показана зависимость КДД от КДО желудочка (а — нормальная зависимость)
Длительное существование гемодинамической перегрузки объемом или сопротивлением (ревматические пороки сердца) или постепенное прогрессирующее снижение сократимости миокарда желудочка (например, при его ремоделировании после перенесенного ИМ или длительном существовании хронической ишемии сердечной мышцы) сопровождается формированием хронической СН (ХСН).
Запомните
В большинстве случаев клинические проявления острой или хронической СН связаны преимущественно с систолической дисфункцией желудочков, которая характеризуется следующими гемодинамическими нарушениями: снижением УО, МО и ФВ; возрастанием КДД (давления наполнения) желудочков; увеличением КДО желудочка (миогенная дилатация); застоем крови в малом или большом круге кровообращения (соответственно, при лево– и правожелудочковой СН).
Примерно в 25–30% случаев в основе развития СН лежат нарушения диастолической функции желудочков. Диастолическая дисфункция развивается при заболеваниях сердца, сопровождающихся нарушением расслабления и наполнения желудочков. Наиболее типичными примерами заболеваний, при которых диастолическая дисфункция проявляется как бы в “чистом” виде, являются аортальный стеноз, ГКМП, экссудативный и констриктивный перикардит, рестриктивные заболевания сердца и др.
Кроме того, диастолическая дисфункция желудочков нередко сочетается с систолической дисфункцией, например, при ИБС или АГ. В этих случаях ухудшение диастолического наполнения обусловлено как увеличением ригидности сердечной мышцы (длительная ишемия миокарда, фиброз, гипертрофия), так и нарушением процессов активного расслабления (снижение энергетического обеспечения, повышение концентрации внутриклеточного Са2+ и т.п.).
Нарушение растяжимости миокарда желудочков приводит к тому, что для обеспечения достаточного диастолического наполнения желудочка кровью и сохранения нормального УО и МО необходимо значительно более высокое давление наполнения, соответствующее более высокому КДД желудочка (см. рис. 1.31). Как видно на рис. 2.1, в, кривая зависимости КДО и КДД при диастолической дисфункции ЛЖ смещается влево и вверх. Поэтому даже небольшой прирост КДО обеспечивается за счет чрезмерно высокого КДД или давления наполнения.
Кроме того, замедление релаксации желудочка приводит к перераспределению диастолического наполнения в пользу предсердного компонента, и значительная часть диастолического кровотока осуществляется не во время фазы быстрого наполнения желудочка, как в норме, а во время активной систолы предсердия. Эти изменения способствуют увеличению давления и размеров предсердия, повышая риск возникновения застоя крови в венозном русле малого или большого круга кровообращения.
Иными словами, диастолическая дисфункция желудочков может сопровождаться клиническими признаками ХСН при нормальной сократимости миокарда и сохраненном сердечном выбросе. При этом полость желудочка обычно остается нерасширенной, поскольку нарушается соотношение КДД и КДО желудочка (рис. 2.1, в).
Запомните
У 25–30% больных хронической СН в основе клинических признаков декомпенсации лежит диастолическая дисфункция желудочков, которая определяет следующие гемодинамические изменения: значительное и раннее повышение КДД (давления наполнения) желудочка; застой крови в венозном русле малого или большого круга кровообращения; малоизмененные или нормальные значения УО и МО; отсутствие значительной дилатации желудочка (малоизмененный КДО).
Следует обратить внимание на то, что во многих случаях ХСН имеет место сочетание систолической и диастолической дисфункции желудочков, что необходимо учитывать при выборе соответствующей медикаментозной терапии.
Из приведенного выше определения СН следует, что этот патологический синдром может развиться не только в результате уменьшения насосной (систолической) функции сердца или его диастолической дисфункции, но и при значительном увеличении метаболических потребностей органов и тканей (гипертиреоз, беременность и т.п.) или при снижении кислородной транспортной функции крови (анемии). В этих случаях МО может оказаться даже повышенным (СН с “высоким МО”), что связано обычно с компенсаторным увеличением ОЦК.
По современным представлениям формирование систолической или диастолической СН тесным образом связано с активацией многочисленных кардиальных и экстракардиальных (нейрогормональных) компенсаторных механизмов. При систолической дисфункции желудочков такая активация вначале носит адаптационный характер и направлена преимущественно на поддержание на должном уровне МО и системного АД. При диастолической дисфункции конечным результатом включения компенсаторных механизмов является повышение давления наполнения желудочков, что обеспечивает достаточный диастолический приток крови к сердцу.
Однако в последующем практически все компенсаторные механизмы трансформируются в патогенетические факторы, способствующие еще большему нарушению систолической и диастолической функции сердца и формированию значительных изменений гемодинамики, характерных для СН.
2.2.2. Кардиальные механизмы компенсации
К числу важнейших кардиальных адаптационных механизмов относятся гипертрофия миокарда и механизм Старлинга.
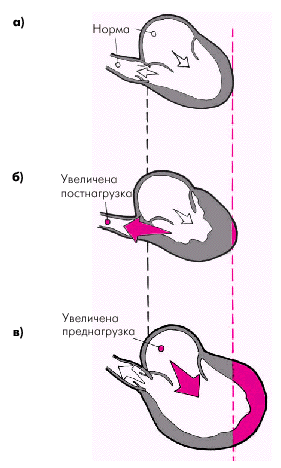
Различают концентрическую и эксцентрическую гипертрофию миокарда желудочков. Длительное хроническое увеличение постнагрузки на какой-либо отдел сердца ведет, как известно, к развитию концентрической гипертрофии миокарда — утолщению мышечной стенки без расширения полости желудочка (рис. 2.2, б). Такая ситуация характерна для артериальной гипертензии, стеноза устья аорты или легочной артерии, для легочной артериальной гипертензии, развивающейся при митральном стенозе, легочном сердце и других заболеваниях. На начальных стадиях этих заболеваний гипертрофия миокарда способствует уменьшению внутримиокардиального напряжения за счет увеличения толщины стенки, позволяя желудочку развивать достаточное внутрижелудочковое давление в систолу.
При другом типе хронической гемодинамической перегрузки сердца — увеличении преднагрузки — развивается эксцентрическая гипертрофия, т.е. тоногенная дилатация полости желудочка, сопровождающаяся умеренной гипертрофией миокарда (рис. 2.2, в). Наиболее часто причинами эксцентрической гипертрофии являются:
недостаточность митрального клапана;
недостаточность клапанов аорты или легочной артерии;
недостаточность трехстворчатого клапана и др.
Рис. 2.2. Концентрическая (б) и эксцентрическая (в) гипертрофия ЛЖ в зависимости от типа хронической перегрузки желудочка (преимущественного увеличения постнагрузки или преднагрузки). а — норма
Умеренная дилатация камер сердца вначале также носит компенсаторный характер, поскольку растяжение кардиомиоцитов, согласно механизму Старлинга, ведет к увеличению силы последующего сокращения и величины выполненной работы (рис. 2.3).
Наконец, при повреждении самой сердечной мышцы (например, при хронической ишемии миокарда) или снижении массы функционирующего миокарда (острый ИМ, постинфарктный кардиосклероз) также развивается гипертрофия сердечной мышцы и так называемая “тоногенная” дилатация ЛЖ, что в течение определенного времени способствует сохранению достаточной величины сердечного выброса.
Рано или поздно компенсаторная реакция сердца на гемодинамическую перегрузку или повреждение миокарда желудочков оказывается недостаточной и происходит снижение сердечного выброса. Так, при гипертрофии сердечной мышцы со временем происходит “изнашивание” сократительного миокарда: истощаются процессы белкового синтеза и энергетического обеспечения кардиомиоцитов, нарушается соотношение между сократительными элементами и капиллярной сетью, повышается концентрация внутриклеточного Са2+, развивается фиброз сердечной мышцы и т.п. Одновременно происходит снижение диастолической податливости камер сердца и развивается диастолическая дисфункция гипертрофированного миокарда. Кроме того, наблюдаются выраженные нарушения метаболизма миокарда:
уменьшается АТФ-азная активность миозина, обеспечивающего сократимость миофибрилл за счет гидролиза АТФ;
нарушается сопряжение возбуждения с сокращением;
нарушается образование энергии в процессе окислительного фосфорилирования и истощаются запасы АТФ и КФ.
Рис. 2.3. Кривая зависимости ударного объема (УО) от исходной длины мышечного волокна (КДО), поясняющая механизм Старлинга.
При нормальной или несколько сниженной сократимости миокарда увеличение КДО сопровождается увеличением УО. Снижение сократимости приводит к нарушению функционирования механизма Старлинга (уплощение кривой)
В результате уменьшается сократимость миокарда, величина МО, возрастает КДД желудочка и появляется застой крови в венозном русле малого или большого круга кровообращения.
Важно помнить, что эффективность механизма Старлинга, обеспечивающего сохранение МО за счет умеренной (“тоногенной”) дилатации желудочка, резко снижается при повышении КДД в ЛЖ больше 18–20 мм рт. ст. Чрезмерное растяжение стенок желудочка (“миогенная” дилатация) сопровождается лишь незначительным увеличением или даже уменьшением силы сокращения, что способствует снижению сердечного выброса.
При диастолической форме СН реализация механизма Старлинга вообще затруднена вследствие ригидности и неподатливости стенки желудочка. Как было показано выше, для достижения должного КДО в этих случаях необходимо очень высокое давление наполнения, КДД желудочка. В результате на самых ранних стадиях диастолической ХСН может развиваться застой крови в малом круге кровообращения.
2.2.3. Экстракардиальные механизмы компенсации
По современным представлениям, основную роль как в процессах адаптации сердца к гемодинамическим перегрузкам или первичному повреждению сердечной мышцы, так и в формировании характерных для СН изменений гемодинамики играет активация нескольких нейроэндокринных систем, важнейшими из которых являются:
симпатико-адреналовая система (САС) и ее эффекторы (адреналин и норадреналин);
ренин-ангиотензин-альдостероновая система (РААС) (почки — надпочечники);
тканевые ренин-ангиотензиновые системы (РАС);
предсердный натрийуретический пептид;
эндотелиальная дисфункция и др.
Гиперактивация симпатико-адреналовой системы
Гиперактивация симпатико-адреналовой системы и повышение концентрации катехоламинов (А и На) является одним из наиболее ранних компенсаторных факторов при возникновении систолической или диастолической дисфункции сердца. Особенно важной оказывается активация САС в случаях развития острой СН. Эффекты такой активации реализуются прежде всего через a- и b-адренергические рецепторы клеточных мембран различных органов и тканей. Основными следствиями активации САС являются (рис. 2.4):
увеличение ЧСС (стимуляция b1-адренергических рецепторов) и, соответственно, МО (поскольку МО = УО х ЧСС);
повышение сократимости миокарда (стимуляция b1- и a1-рецепторов);
системная вазоконстрикция и повышение ОПСС и АД (стимуляция a1-рецепторов);
повышение тонуса вен (стимуляция a1-рецепторов), что сопровождается увеличением венозного возврата крови к сердцу и увеличением преднагрузки;
стимуляция развития компенсаторной гипертрофии миокарда;
активирование РААС (почечно-надпочечниковой) в результате стимуляции b1-адренергических рецепторов юкстагломерулярных клеток и тканевых РАС за счет дисфункции эндотелия.
Таким образом, на начальных этапах развития заболевания повышение активности САС способствует увеличению сократимости миокарда, притока крови к сердцу, величины преднагрузки и давления наполнения желудочков, что в конечном итоге приводит к сохранению в течение определенного времени достаточного сердечного выброса. Однако длительная гиперактивация САС у больных хронической СН может иметь многочисленные негативные последствия, способствуя:
1. Значительному увеличению преднагрузки и постнагрузки (за счет чрезмерной вазоконстрикции, активации РААС и задержки натрия и воды в организме).
2. Повышению потребности миокарда в кислороде (в результате положительного инотропного эффекта активации САС).
3. Уменьшению плотности b-адренергических рецепторов на кардиомиоцитах, что со временем приводит к ослаблению инотропного эффекта катехоламинов (высокая концентрация катехоламинов в крови уже не сопровождается адекватным увеличением сократимости миокарда).
4. Прямому кардиотоксическому эффекту катехоламинов (некоронарогенные некрозы, дистрофические изменения миокарда).
5. Развитию фатальных желудочковых нарушений ритма (желудочковой тахикардии и фибрилляции желудочков) и т.д.
Рис. 2.4. Эффекты гиперактивации симпатико-адреналовой системы (САС) при систолической ХСН. Объяснение в тексте
Гиперактивация ренин-ангиотензин-альдостероновой системы
Гиперактивация РААС играет особую роль в формировании СН. При этом имеет значение не только почечно-надпочечниковая РААС с циркулирующими в крови нейрогормонами (ренином, ангиотензином-II, ангиотензином-III и альдостероном), но и локальные тканевые (в том числе миокардиальная) ренин-ангиотензиновые системы.
На рис. 2.5. показана упрощенная схема РААС. Активация почечной ренин-ангиотензиновой системы, наступающая при любом самом незначительном снижении перфузионного давления в почках, сопровождается выделением клетками ЮГА почек ренина, расщепляющего ангиотензиноген с образованием пептида — ангиотензина I (АI). Последний под действием ангиотензин-превращающего фермента (АПФ) трансформируется в ангиотензин II, который является основным и наиболее мощным эффектором РААС. Характерно, что ключевой фермент этой реакции — АПФ — локализуется на мембранах эндотелиальных клеток сосудов легких, проксимальных канальцев почек, в миокарде, плазме, где и происходит образование АII. Его действие опосредуется специфическими ангиотензиновыми рецепторами (АТ1 и АТ2), которые находятся в почках, сердце, артериях, надпочечниках и т.д. Важно, что при активации тканевых РАС имеются и другие пути (помимо АПФ) превращения АI в АII: под действием химазы, химазоподобного фермента (CAGE), катепсина G, тканевого активатора плазминогена (ТАП) и др.
Рис. 2.5. Упрощенная схема ренин-ангиотензин-альдостероновой системы (РААС) и основные эффекты ее гиперактивации при ХСН. Объяснения и обозначения в тексте
Наконец, воздействие АII на АТ2-рецепторы клубочковой зоны коркового вещества надпочечников приводит к образованию альдостерона, основным эффектом которого является задержка в организме натрия и воды, что способствует увеличению ОЦК.
В целом активация РААС сопровождается следующими эффектами:
выраженной вазоконстрикцией, повышением АД;
задержкой в организме натрия и воды и увеличением ОЦК;
инициированием развития гипертрофии и ремоделирования сердца;
активацией образования соединительной ткани (коллагена) в миокарде;
повышением чувствительности миокарда к токсическому влиянию катехоламинов.
Активация РААС при острой СН и на начальных этапах развития хронической СН имеет компенсаторное значение и направлена на поддержание нормального уровня АД, ОЦК, перфузионного давления в почках, увеличение пред- и постнагрузки, увеличение сократимости миокарда. Однако в результате длительной гиперактивации РААС развивается ряд отрицательных эффектов:
1. увеличение ОПСС и снижение перфузии органов и тканей;
2. чрезмерное увеличение постнагрузки на сердце;
3. значительная задержка жидкости в организме, что способствует формированию отечного синдрома и повышению преднагрузки;
4. инициация процессов ремоделирования сердца и сосудов, в том числе гипертрофии миокарда и гиперплазии гладкомышечных клеток;
5. стимуляция синтеза коллагена и развитие фиброза сердечной мышцы;
6. развитие некроза кардиомиоцитов и прогрессирующее повреждение миокарда с формированием миогенной дилатации желудочков;
7. повышение чувствительности сердечной мышцы к катехоламинам, что сопровождается возрастанием риска возникновения фатальных желудочковых аритмий у больных СН.
Система аргинин-вазопрессин (антидиуретический гормон)
Антидиуретический гормон (АДГ), секретируемый задней долей гипофиза, участвует в регуляции проницаемости для воды дистальных отделов канальцев почек и собирательных трубок. Например, при недостатке в организме воды и дегидратации тканей происходит уменьшение объема циркулирующей крови (ОЦК) и увеличение осмотического давления крови (ОДК). В результате раздражения осмо- и волюморецепторов усиливается секреция АДГ задней долей гипофиза. Под влиянием АДГ повышается проницаемость для воды дистальных отделов канальцев и собирательных трубок, и, соответственно, усиливается факультативная реабсорбция воды в этих отделах. В итоге выделяется мало мочи с высоким содержанием осмотически активных веществ и высокой удельной плотностью мочи.
Наоборот, при избытке воды в организме и гипергидратации тканей в результате увеличения ОЦК и уменьшения ОДК происходит раздражение осмо- и волюморецепторов, и секреция АДГ резко снижается или даже прекращается. В результате реабсорбция воды в дистальных отделах канальцев и собирательных трубках снижается, тогда как Na+ продолжает реабсорбироваться в этих отделах. Поэтому выделяется много мочи с низкой концентрацией осмотически активных веществ и низкой удельной плотностью.
Нарушение функционирования этого механизма при сердечной недостаточности может способствовать задержке воды в организме и формированию отечного синдрома. Чем меньше сердечный выброс, тем больше раздражение осмо- и волюморецепторов, что приводит к увеличению секреции АДГ и, соответственно, задержке жидкости.
Предсердный натрийуретический пептид
Предсердный натрийуретический пептид (ПНУП) является своеобразным антагонистом вазоконстрикторных систем организма (САС, РААС, АДГ и других). Он продуцируется миоцитами предсердий и выделяется в кровоток при их растяжении. ПНУП вызывает вазодилатирующий, натрийуретический и диуретический эффекты, угнетает секрецию ренина и альдостерона.
Секреция ПНУП — это один из наиболее ранних компенсаторных механизмов, препятствующих чрезмерной вазоконстрикции, задержке Nа+ и воды в организме, а также увеличению пред- и постнагрузки.
Активность ПНУП быстро усиливается по мере прогрессирования СН. Однако, несмотря на высокий уровень циркулирующего ПНУП, степень его положительных эффектов при хронической СН заметно снижается, что связано, вероятно, с уменьшением чувствительности рецепторов и увеличением расщепления пептида. Поэтому максимальный уровень циркулирующего ПНУП ассоциируется с неблагоприятным течением хронической СН.
Нарушения эндотелиальной функции
Нарушениям эндотелиальной функции в последние годы придается особое значение в формировании и прогрессировании ХСН. Дисфункция эндотелия, возникающая под действием различных повреждающих факторов (гипоксии, чрезмерной концентрации катехоламинов, ангиотензина II, серотонина, высокого уровня АД, ускорения кровотока и т.д.), характеризуется преобладанием вазоконстрикторных эндотелийзависимых влияний и закономерно сопровождается повышением тонуса сосудистой стенки, ускорением агрегации тромбоцитов и процессов пристеночного тромбообразования (см. главу 1).
Напомним, что к числу важнейших эндотелийзависимых вазоконстрикторных субстанций, повышающих сосудистый тонус, агрегацию тромбоцитов и свертываемость крови, относятся эндотелин-1 (ЭТ1), тромбоксан А2, простагландин PGH2, ангиотензин II (АII) и др.
Они оказывают существенное влияние не только на сосудистый тонус, приводя к выраженной и стойкой вазоконстрикции, но и на сократимость миокарда, величину преднагрузки и постнагрузки, агрегацию тромбоцитов и т.д. (подробнее см. главу 1). Важнейшим свойством эндотелина-1 является его способность “запускать” внутриклеточные механизмы, приводящие к усилению белкового синтеза и развитию гипертрофии сердечной мышцы. Последняя, как известно, является важнейшим фактором, так или иначе осложняющим течение СН. Кроме того, эндотелин-1 способствует образованию коллагена в сердечной мышце и развитию кардиофиброза. Существенную роль вазоконстрикторные субстанции играют в процессе пристеночного тромбообразования (рис. 2.6).
Показано, что при тяжелой и прогностически неблагоприятной ХСН уровень эндотелина-1 повышен в 2–3 раза. Его концентрация в плазме крови коррелирует с выраженностью нарушений внутрисердечной гемодинамики, давлением в легочной артерии и уровнем летальности у пациентов с ХСН.
Запомните
1. Одним из ведущих патогенетических механизмов формирования и прогрессирования сердечной недостаточности является гиперактивация нейрогормональных систем организма — САС, РААС, АДГ, ПНУП и др., а также дисфункция эндотелия. 2. На начальных этапах развития заболевания активация этих систем, возникающая в результате систолической или диастолической дисфункции сердца, носит адаптационный характер и направлена на сохранение достаточного сердечного выброса, системного АД и перфузии органов и тканей. Этот эффект реализуется благодаря: увеличению ЧСС; повышению сердечного выброса за счет гиперфункции с последующей гипертрофией; увеличению постнагрузки (вазоконстрикция); увеличению преднагрузки и ОЦК (физиологическая задержка натрия и воды) и др. 3. Длительная чрезмерная активация нейрогормональных систем приводит к: избыточной задержке натрия и воды в организме (отечный синдром); резкому увеличению ОПСС (нарушение перфузии органов и тканей); чрезмерному возрастанию пред- и постнагрузки, что ведет к снижению функции сердца; стимулированию синтеза коллагена и развитию кардиофиброза; развитию некрозов кардиомиоцитов, прогрессирующему повреждению сердечной мышцы и формированию миогенной дилатации сердца.
Рис. 2.6. Роль дисфункции эндотелия в формировании и прогрессировании ХСН. Объяснения и обозначения в тексте. На — норадреналин
Таким образом, описанные эффекты гиперактивации нейрогормональных систем вместе с типичными нарушениями гемодинамики лежат в основе характерных клинических проявлений СН. Причем, симптоматика острой СН главным образом определяется внезапно наступившими расстройствами гемодинамики (выраженным снижением сердечного выброса и ростом давления наполнения), микроциркуляторными нарушениями, которые усугубляются активацией САС, РААС (преимущественно почечной).



 Запомните
Наиболее частыми причинами сердечной недостаточности являются: ИБС, включая острый ИМ и постинфарктный кардиосклероз; артериальная гипертензия, в том числе в сочетании с ИБС; клапанные пороки сердца.
Запомните
Наиболее частыми причинами сердечной недостаточности являются: ИБС, включая острый ИМ и постинфарктный кардиосклероз; артериальная гипертензия, в том числе в сочетании с ИБС; клапанные пороки сердца.