 |


Селективность разделения в газовой хроматографии
|
|
|
|
Закономерности газохроматографической селективности.
1. Вариант газо-жидкостной хроматографии. Согласно закону Рауля, упругость пара растворенного вещества над раствором пропорциональна его мольной доле в растворе:
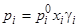 , (6.1)
, (6.1)
где 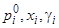 — соответственно: давление насыщенных паров чистого аналита, его доля и коэффициент активности в НФ. Величина
— соответственно: давление насыщенных паров чистого аналита, его доля и коэффициент активности в НФ. Величина 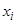 пропорциональна концентрации аналита в НФ, поэтому для константы Генри запишем:
пропорциональна концентрации аналита в НФ, поэтому для константы Генри запишем:
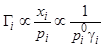 (6.2)
(6.2)
— удерживание обратно пропорционально давлению насыщенных паров чистого аналита. Связь (6.2), записанная для отношения коэффициентов распределения двух веществ, называется уравнением Херингтона.
В свою очередь, давление насыщенных паров при некоторой температуре T связано с температурой кипения жидкого аналита и соответствующим давлением (которое в открытой системе равно атмосферному давлению  ):
):
 , (6.3)
, (6.3)
где 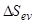 — энтропия испарения,
— энтропия испарения, 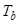 — температура кипения, R — газовая постоянная. Комбинируя приведенные связи, для коэффициента селективности (17) близких по свойствам аналитов получим простое соотношение:
— температура кипения, R — газовая постоянная. Комбинируя приведенные связи, для коэффициента селективности (17) близких по свойствам аналитов получим простое соотношение:
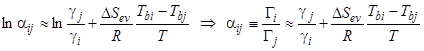 , (6.4)
, (6.4)
где — средняя величина энтропии испарения для рассматриваемых аналитов; правое выражение получено при использовании приближенного равенства  для
для 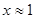 . Применяя правило Трутона, согласно которому мольная энтропия испарения неполярной жидкости составляет величину порядка 90 Дж/(моль∙К), получим величину коэффициента в (6.4) около 11. В полученную формулу входит обратное отношение коэффициентов активностей, зависящих от силы взаимодействия аналита с НФ: величина коэффициента активности тем ниже, чем сильнее такое взаимодействие. Итак, если полярное вещество «i» и неполярное вещество «j» имеют одинаковые температуры кипения (в общем случае — одинаковые произведения температур кипения на энтропии испарения), то получим в условиях ГХ:
. Применяя правило Трутона, согласно которому мольная энтропия испарения неполярной жидкости составляет величину порядка 90 Дж/(моль∙К), получим величину коэффициента в (6.4) около 11. В полученную формулу входит обратное отношение коэффициентов активностей, зависящих от силы взаимодействия аналита с НФ: величина коэффициента активности тем ниже, чем сильнее такое взаимодействие. Итак, если полярное вещество «i» и неполярное вещество «j» имеют одинаковые температуры кипения (в общем случае — одинаковые произведения температур кипения на энтропии испарения), то получим в условиях ГХ:
|
|
|
· на полярной НФ: 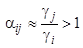 — более высокую селективность к полярному веществу «i»;
— более высокую селективность к полярному веществу «i»;
· на неполярной НФ: 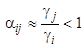 — более высокую селективность к неполярному веществу «j».
— более высокую селективность к неполярному веществу «j».
Если оба вещества имеют одинаковую полярность, то отношение коэффициентов активностей можно положить равным 1, тогда из (6.4) следует оценочное соотношение:
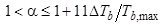 , (6.5)
, (6.5)
откуда следует, что
· удерживается сильнее то вещество, которое имеет б о льшую температуру кипения;
· каждый градус различия в температурах кипения добавляет к коэффициенту селективности разделения подобных веществ величину, примерно равную 11-кратной обратной температуре кипения (в шкале Кельвина); в диапазоне температур, наиболее часто используемых в ГХ (100-200оС), эта величина не превышает 0.03. В рассмотренном в конце главы 3 примере найдено, что для разделения пары веществ с  эффективность системы должна составлять 25000 теоретических тарелок. Приведенный выше вывод позволяет оценить в качестве такой пары веществ неполярные соединения, различающиеся по температуре кипения на 1 градус. Согласно формуле (52), при росте
эффективность системы должна составлять 25000 теоретических тарелок. Приведенный выше вывод позволяет оценить в качестве такой пары веществ неполярные соединения, различающиеся по температуре кипения на 1 градус. Согласно формуле (52), при росте  в 3 раза (например, для пары веществ, различающихся по температуре кипения на 3 градуса), требующаяся эффективность колонки снижается на порядок, становясь легко реализуемой величиной.
в 3 раза (например, для пары веществ, различающихся по температуре кипения на 3 градуса), требующаяся эффективность колонки снижается на порядок, становясь легко реализуемой величиной.
2. Вариант газоадсорбционной хроматографии. Существуют различные модели адсорбции: они базируются либо на подробном рассмотрении сложных явлений межмолекулярного взаимодействия с целью априорного расчета хроматографического удерживания по структуре молекул и адсорбента, либо на привлечении аппарата описания аналогичных систем, но являющихся более изученными. Очевидно, что второй тип моделей адсорбции проще, однако их предсказательная сила слабее. Например, в рамках известной потенциальной теории Поляни существует представление об адсорбированном слое, как о двумерной жидкости, находящейся в контакте с газовой фазой [10]. Для такой модели адсорбции возможно описание, аналогичное представленному выше для газожидкостного варианта. Отличие количественных соотношений будет только в учете поправок, отражающих наличие вместо трехмерной пленки — двумерной «пленки неподвижной фазы», имитирующей адсорбент. Тем не менее, общий вид закономерностей в рамках этой модели останется прежним.
|
|
|
Таким образом, закономерности селективности для различных вариантов ГХ имеют схожий характер и поэтому часто нет необходимости уточнять, является ли механизм разделения распределительным или адсорбционным.
Полярность и селективность неподвижной фазы.
Полярность неподвижной фазы в ГХ оценивается по отношению к условно выбранным эталонам. За точку отсчета шкалы полярности НФ принимается сквалан, а за 100 — β,β-оксидипропионитрил (бис-цианоэтиловый эфир). Полярность НФ с индексом «x» определяется по формуле:
 , (6.6)
, (6.6)
где 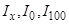 - индексы Ковача «эталонного» соединения для соответствующих фаз.
- индексы Ковача «эталонного» соединения для соответствующих фаз.
Более тонкая характеристика – селективность НФ, включающая в себя оценку вкладов всего спектра взаимодействий в адсорбции, может быть найдена по методу Роршнайдера. НФ описывается вектором, состоящим из 5 компонент: 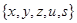 ; за точку отсчета принимается сквалан с нулевым вектором:
; за точку отсчета принимается сквалан с нулевым вектором:  . Для процедуры тестирования выбраны 5 соединений: бензол, этанол, метилэтилкетон, нитрометан и пиридин, — полярность которых характеризуется вектором
. Для процедуры тестирования выбраны 5 соединений: бензол, этанол, метилэтилкетон, нитрометан и пиридин, — полярность которых характеризуется вектором 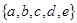 (таблица 6.3).
(таблица 6.3).
Таблица 6.3. Эталоны для определения селективности НФ в ГХ
| Соединение (аналит) | Вектор полярности аналита |
| Бензол | {1,0,0,0,0} |
| Этанол | {0,1,0,0,0} |
| Метилэтилкетон | {0,0,1,0,0} |
| Нитрометан | {0,0,0,1,0} |
| Пиридин | {0,0,0,0,1} |
Вместо перечисленных веществ для тестирования НФ могут быть использованы другие аналиты, векторы которых имеют нецелые компоненты.
Селективность НФ определяется из уравнения:
 (6.7)
(6.7)
Например, компоненту x можно найти из сравнения индексов Ковача бензола, полученных из опытов на рассматриваемом адсорбенте и на сквалане; для компоненты y нужно провести опыты с этанолом и т.д. Для полной характеристики адсорбента необходимо решить систему из пяти уравнений (6.7), полученных для пяти соединений из разных гомологических рядов, различающихся не столько полярностью, сколько вкладом в их адсорбцию различных типов взаимодействий. Из формы уравнения (6.7) следует, что в список тестовых веществ не могут быть включены алканы, так как для них левая часть уравнения тождественно равна нулю.
|
|
|
Характеристика некоторых распространенных в ГХ фаз дана в таблице 6.4.
Таблица 6.4. Коэффициенты Роршнайдера для НФ в ГХ
| Неподвижная фаза | x | y | z | u | s |
| Силикон OV-1, OV-101 | 0.16 | 0.20 | 0.50 | 0.85 | 0.48 |
| Апиезон L | 0.32 | 0.39 | 0.25 | 0.48 | 0.55 |
| Силикон OV-3 | 0.42 | 0.81 | 0.85 | 1.52 | 0.89 |
| Силикон OV-11 | 1.13 | 1.57 | 1.69 | 2.66 | 1.95 |
| Твин 80 | 2.14 | 4.20 | 2.78 | 5.20 | 3.65 |
| Этиленгликольизофталат, карбовакс 4000 | 3.1 | 5.1 | 4.0 | 6.8 | 5.2 |
| Цианэтилсахароза, β,β-оксидипропионитрил | 5.6 | 8.6 | 7.7 | 11.6 | 9.0 |
Мак-Рейнольдс построил свою систему классификации НФ [11], используя ту же идею, но привлекая расширенный до десяти набор тестовых соединений, взятых из разных классов органических веществ.
Детекторы
Детектирующая система газового хроматографа может иметь одновременно несколько детекторов: один универсальный и ряд селективных детекторов. В качестве универсальных детекторов в ГХ используют детекторы теплопроводности, пламенно-ионизационные и масс-спектрометрические детекторы. Для ряда специальных задач, связанных с определением микроэлементных примесей – своеобразных «отпечатков», несущих информацию о происхождении объекта (нефти, сельскохозяйственной культуры и т.п.), применяются селективные детекторы. Основные детекторы, использующиеся в ГХ, перечислены в таблице 6.5.
Таблица 6.5. Детекторы для газовой хроматографии
| Название детектора | Избирательность | Предел обнаружения | Диапазон линейности (число десятичных порядков) |
| ДТП детектор теплопровод-ности (катарометр) | неизбирателен | 10 нг/мл | |
| ПИД пламенно-ионизацион-ный детектор | неизбирателен для органики | 0.1 пг/с | |
| ДЭЗ электронно-захватный детектор | наличие галогенов, электроотрицатель-ных групп | 0.05 пг/с | 4.5-5 |
| ТИД термоионный детектор | наличие фосфора, азота | 1 фг/с, 10 фг/с | |
| ФИД фотоионизационный детектор | универсален, избирателен к ароматике | 0.2 мкг/л | |
| ПФД пламенно-фотометри-ческий детектор | наличие фосфора, серы | 0.3 пг/с 20 пг/с | |
| МС масс-спектрометр | неизбирателен по полному току | 1 пг | ~5 |
Примечание. 1 нг=1000 пг=1000000 фг.
|
|
|
Первым в аналитической газовой хроматографии стали применять детектор теплопроводности (катарометр) в силу следующих его достоинств: универсальность (т.е. неизбирательность) и приемлемая чувствительность, легкая миниатюризация и герметичность ячейки детектора, простота изготовления детектора, хорошая линейность сигнала и непрерывность измерения. Схема ячейки катарометра представлена на рис. 6.2.
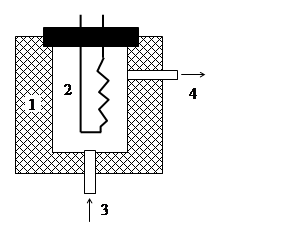
Рис. 6.2. Схема ячейки катарометра: корпус (1), нить накаливания (2), вход ПФ после колонки (3), сброс ПФ (4).
Принцип детектирования заключается в использовании потока газа-носителя в качестве проводника тепла между нагретой нитью и термостатированным при небольшой температуре металлическим корпусом ячейки. Чем выше теплопроводность газа, тем сильнее снижается температура нити. Способ детектирования заключается в измерении тока, протекающего через нагретую спираль, сопротивление которой зависит от ее температуры (как и для любого проводника – с повышением температуры сопротивление спирали растет, а, следовательно, ток при постоянном напряжении падает). Для получения максимальных эффектов в качестве газа-носителя используют водород или гелий – газы с максимальной теплопроводностью, которая в 6-10 раз выше теплопроводности большинства аналитов в ГХ. Наиболее распространенной схемой включения катарометра является разностная схема с двумя ячейками, одна из которых (ячейка сравнения) включена в поток чистого газа-носителя, не содержащего аналитов. Как видно из таблицы 6.5, чувствительность катарометра ниже, а диапазон линейности уже, чем у других типов детекторов. К недостаткам этого принципа детектирования можно отнести также необходимость применения только легких газов-носителей (водород, гелий). Достоинством детекторов теплопроводности является то, что в результате детектирования компоненты пробы, как правило, не разрушаются.
Наиболее распространенным в ГХ является пламенно-ионизационный детектор (ПИД). Его схема приведена на рис. 6.3. В корпусе 1 ячейки расположена кислородно-водородная горелка 6, каналы для ввода и вывода газов 2,3,4,7 и система электродов 5. Верхний электрод заряжен положительно и называется коллектором.
|
|
|

Рис. 6.3. Схема пламенно-ионизационного детектора: корпус (1), вход ПФ после колонки (2), подача водорода (3), подача кислорода (4), электроды (5), пламя (6), выход газов (7).
В основе работы ПИД лежит увеличение электропроводности водородного пламени в присутствии органических веществ, которые поступают в пламя с потоком газа-носителя из колонки. При сгорании веществ, содержащих СН-группы, образуются ионы и свободные электроны согласно реакции:
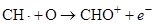 .
.
ПИД позволяет детектировать любые вещества, содержащие С-С и С-Н – связи. К карбонильным, спиртовым, аминным группам, галогенам детектор менее чувствителен. Полностью окисленные формы с его помощью определять нельзя.
Сигнал ПИД пропорционален массе вещества, проходящей через ячейку в единицу времени. Чувствительность и диапазон линейности детектора чрезвычайно высоки (см. табл. 6.5).
Детектор электронного захвата действует по принципу газоразрядного счетчика. В специальной ячейке с двумя электродами газ, поступающий после колонки, подвергается облучению потоком  -частиц, испускаемых изотопом 63Ni или тритием. Газ ионизируется и в отсутствие аналитов в ячейке создается постоянный ток, переносимый, главным образом, свободными электронами, обладающими высокой подвижностью. Аналиты, содержащие электроотрицательные заместители: галогены, хиноны, нитрогруппы, - способны захватывать электроны с образованием стабильных анионов, тем самым способствуя уменьшению электронного тока в ячейке. По отношению к другим аналитам ДЭЗ нечувствителен. ДЭЗ имеет высокую чувствительность и очень полезен при определении токсичной хлорорганики, например, пестицидов.
-частиц, испускаемых изотопом 63Ni или тритием. Газ ионизируется и в отсутствие аналитов в ячейке создается постоянный ток, переносимый, главным образом, свободными электронами, обладающими высокой подвижностью. Аналиты, содержащие электроотрицательные заместители: галогены, хиноны, нитрогруппы, - способны захватывать электроны с образованием стабильных анионов, тем самым способствуя уменьшению электронного тока в ячейке. По отношению к другим аналитам ДЭЗ нечувствителен. ДЭЗ имеет высокую чувствительность и очень полезен при определении токсичной хлорорганики, например, пестицидов.
Термоионный детектор по своему устройству похож на ПИД. Отличие состоит в том, что горючая смесь сильно обеднена водородом и не способна к самостоятельному горению. Кроме того, в качестве отрицательного (нижнего) электрода используется платиновая проволока, на которую помещена капля рубидиевого стекла. В рабочем состоянии проволока раскалена; вокруг раскаленного стекла образуется слой плазмы, попадая в который азот- и фосфор-содержащие молекулы образуют радикалы  , которые реагируют с атомами рубидия с образованием ионов. Катионы рубидия притягиваются отрицательным электродом и снова оказываются в стекле, а цианид- и фосфит-ионы направляются к положительному коллектору, создавая электрический ток.
, которые реагируют с атомами рубидия с образованием ионов. Катионы рубидия притягиваются отрицательным электродом и снова оказываются в стекле, а цианид- и фосфит-ионы направляются к положительному коллектору, создавая электрический ток.
Таким образом, ТИД обладает селективностью к азот- и фосфор-органике: его чувствительность к таким веществам в 10000 раз выше, чем к углеводородам.
Принцип действия фотоионизационного детектора (ФИД) заключается в ионизации молекул, элюируемых с хроматографической колонки, в вакууме под действием УФ-излучения и измерении возникающего ионного тока. Изменяя энергию излучения, можно варьировать чувствительность детектирования соединений различных классов. Особенно низкий предел обнаружения у ФИД для ароматических углеводородов (при использовании лампы с энергией 10.2 эВ). Положительной особенностью ФИД является то, что он не разрушает детектируемые соединения, и его можно использовать в комбинации с другими детекторами для более надежной идентификации сложных смесей.
Пламенно-фотометрический детектор используется для селективного детектирования фосфор- и серо-содержащей органики. Газ, выходящий из колонки, попадает в водородно-воздушное пламя. В результате неполного сгорания фосфор- и серо-содержащих веществ образуются возбужденные формы, которые испускают излучение. Для его регистрации используют фотоэлектронный умножитель (ФЭУ). Фосфор детектируют при 526 нм, а серу – при 394 нм.
Масс-спектрометр используется в гибридных методах хромато-масс-спектрометрии — наиболее мощных средствах анализа объектов органической природы.
Первоначальные попытки объединения газовой хроматографии и масс-спектрометрии были продиктованы стремлением добиться высокой чувствительности детектирования. Однако в дальнейшем богатые возможности масс-спектрометрии как средства изучения структуры соединений, разделенных с помощью газовой хроматографии, были широко использованы в многочисленных конструкциях комбинированных приборов, объединяющих преимущества обоих методов.
Основная идея этого объединения заключается в том, чтобы зафиксировать масс-спектр идентифицируемого соединения за время, малое по сравнению с временем, в течение которого соответствующая зона выходит из капиллярной колонки. Разделенные в колонке соединения поступают в масс-спектрометр, где их молекулы ионизируются и образующиеся ионы подвергаются разделению по их массе, точнее по величине отношения массы к заряду. В процессе ионизации органических соединений обычно происходит распад исходной молекулы на более простые фрагменты, регистрируемые как ионы с меньшей массой. Ионизация молекул может осуществляться при соударениях с быстро движущимися электронами, термическим путем, под влиянием высокочастотного электромагнитного поля, при действии искрового или дугового разряда или в результате химической ионизации. Образующиеся ионы разгоняются в электрическом поле, после чего подвергаются разделению по отношению их массы к заряду с помощью анализаторов различного типа.
Наиболее высокой разрешающей способностью обладают спектрометры, в которых дифференциация ионов происходит за счет различного отклонения их в электрических и магнитных полях — вследствие зависимости угла отклонения от отношения массы к заряду иона (т/z). Для регистрации сфокусированного пучка ионов в настоящее время чаще всего применяют электрометрический метод. Изменяя напряженность электрического или магнитного поля отклоняющей системы, на электроды коллектора — детектора ионов — направляют пучки сфокусированных частиц с последовательно увеличивающейся величиной отношения т/z. Запись значений тока коллектора в зависимости от напряжения отклоняющего поля представляет собой масс-спектр анализируемого соединения.
Гораздо более, чем дорогостоящие и громоздкие приборы классического типа, распространены масс-спектрометры, в которых ионы с разным отношением массы к заряду различают по времени, в течение которого они проходят определенный путь в вакууме. Такие времяпролетные масс-спектрометры проще по устройству, компактнее и дешевле. Они обладают очень высоким быстродействием, так что весь масс-спектр изучаемого соединения может быть зарегистрирован в доли секунды. Однако разрешающая способность и чувствительность таких приборов заметно ниже, чем у масс-спектрометров с электрическим или электромагнитным отклонением ионного пучка. В ряде конструкций масс-спектрометров для разделения ионов по величине отношения массы к заряду иона используют закономерности движения заряженных частиц в быстро меняющемся электрическом поле, создаваемом системой из четырех электродов определенной конфигурации. Такие приборы, называемые квадрупольными масс-спектрометрами, обладают несколько меньшей разрешающей способностью.
В большинстве случаев в ионном источнике органические молекулы подвергаются воздействиям, энергия которых заметно превышает энергию связей. В наиболее распространенных масс-спектрометрах, в которых ионизация осуществляется путем соударения молекул с быстро движущимися электронами, энергия последних достигает 70 эВ, что намного превышает не только ионизационный потенциал углерода (11.4 эВ), но и энергию самых прочных связей, встречающихся в органических соединениях. В масс-спектре органических веществ обычно, кроме молекулярного иона, возникающего при отрыве электрона от всей молекулы, наблюдают сигналы частиц с меньшей массой, называемых осколочными ионами. Последние возникают в результате разрыва химических связей в анализируемом соединении и отщепления от него отдельных радикалов и групп атомов.
Сопоставление величин массы и интенсивности сигналов наблюдаемых ионов в большинстве случаев позволяет делать вполне определенные заключения о химической структуре изучаемых соединений. Так, например, если масс-спектр соединения содержит пики с т/z, равным 168 (4.4), 139 (38), 125 (14), 97 (18), с относительной интенсивностью, указанной в скобках (в процентах), то можно сделать однозначный выбор между двумя изомерными структурами (I и II), показанными на рис. 6.4, на основании следующих соображений (цит. по [12]).
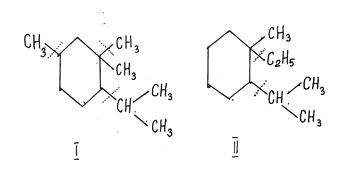
Рис. 6.4. Возможные схемы фрагментации молекул двух изомеров (I и II) алкилзамещенного циклогексана
Указанные значения т/z ионов (139 и 125) соответствуют отщеплению этильной и изопропильной групп. Пик, возникающий при отщеплении метильной группы (т/z =153), отсутствует. Зато в спектре имеется интенсивный пик с т/z =97, который может отвечать отщеплению этилена (т = 28) от иона с т/z =125 или пропилена (m = 42) от иона с т/z =139 при их последующей перегруппировке. Таким образом, масс-спектр однозначно указывает на то, что анализируемое соединение имеет структуру II.
|
|
|
