 |


Потенциометрия, как метод физико-химического анализа.
|
|
|
|
1. Согласно нижеприведенным уравнениям измеряя ЭДС можно получить:
∆G = - zFE. (1.21)
T ∆S = zFT (dE /dT). (2.21)
∆H = - zFE + zFT (dE /dT). (3.21)
2. Константу равновесия.
E0 = RT/zF ln Ka (4.21)
ln Ka = E0zF / RT. (5.21)
3. Произведения растворимости соли.
- Ag, AgClï KCl (aq) ïï KCl (aq) ïHg2Cl2, Hg +
Положим нам необходимо определить растворимость хлорида серебра. Для начала необходимо определить его электродный потенциал из ЭДС цепи.
Затем мы представляем реакцию:
AgCl + l- → Ag + Cl- (6.21)
Как
AgCl → Ag+ + Cl- (7.21)
(7.21) - это равновесная реакция потенциала не дает.
Ag+ + l- → Ag (8.21)
(8.21) - потенциало-образующая реакция.
Электродный потенциал реакции (6.21):
E1 = E10 - RT/zF ln a(Cl-)
Реакции (8.21):
E2 = E20 + RT/zF ln a (Ag +)
E1 = E2
Поэтому:
E10 - RT/zF ln a(Cl-) = E20 + RT/zF ln a Ag +/ a Ag
ln a(Cl-) a (Ag +) = zF / RT (E10 - E20)
где ПР = a(Cl-) a (Ag +).
4. Аналогично можно получить константу нестойкости комплексного соединения.
5. Потенциометрия используется также для определения активностей веществ в твердых растворах (сплавах).
Основные понятия химической кинетики (скорость, константа скорости, порядок реакции, молекулярность, энергия активации, катализаторы)
Химическая кинетика – это учение о скоростях химических процессов, механизмах превращений и закономерностях их протекания во времени.
Эта наука позволяет предсказать, с какой скоростью осуществится процесс. Но ответ на данный вопрос требует знание о структуре, механизме явления, здесь не достаточно сведений о начальном и конечном состоянии системы как в термодинамике. Поэтому получение сведений в кинетике более трудоемкий процесс.
Химические реакции сложны и протекают через множества элементарных стадий.
Механизм реакции – совокупность элементарных актов.
|
|
|
Любое элементарное превращение – обратимо. Механизм реакции, как правило, не известен, так как образующиеся промежуточные вещества очень нестабильны. Реакция может состоять из множества последовательных и параллельных актов со многими промежуточными частицами.
Пример: H2 + I2 ↔ 2HI, на деле I2 ↔ 2I., I. + H2 ↔ HI + H., H. + I2 ↔ HI + I. ….
Молекулярность элементарной стадии - число частиц участвующих в акте (не более трех).
Скорость химической реакции – изменение количества какого-либо компонента за единицу времени в единице реакционного пространства.
V = (1/R)(dni/dt) (1.22)
Где R – площадь или объем.
Основной закон кинетики:
Гольдберг и Ваге открыли что:
V = kc1n1c2n2 (2.22)
k – константа скорости реакции; n1, n2, - стехиометрические коэффициенты в уравнении реакции их сумма порядок реакции. Так как реакции проходят по сложным механизмам, порядок по реакции редко совпадает с действительным порядком. Порядок может быть нулевым, первой степени, второй, дробным.
Приравнивая (1.22) и (2.22) получаем уравнение, которое можно решить относительно концентраций и времени. Поставив эксперимент можно найти эмпирическую зависимость концентрации от времени.
Катализатор – вещество, которое ускоряет ход химической реакции, посредством образовании активированного комплекса в одной или нескольких элементарных стадий химической реакции.
Ингибитор – замедляет реакцию.
Катализатор не смещает равновесия химической реакции, не влияет на ее константу, а лишь приближает момент наступления равновесия.
Катализ может быть гомогенным и гетерогенным.
Гетерогенный катализатор и реагенты находятся в разных фазах. Реакция протекает на поверхности раздела. Здесь прежде всего надо отметить в качестве катализаторов благородные элементы. У подобных элементов много электронов на верхних энергетических уровнях, множество связывающих и разрыхляющих орбиталей в результате этого возможно образование различных активированных комплексов, где реагенты будут лигандами. При этом связи внутри соединений, за счет перераспределения электронной плотности на связь с катализатором ослабляются. Вследствие чего уменьшается количество энергии необходимой для перестройки этих связей. С течением времени структура катализатора и состав может меняться, в результате взаимодействия с реагентами и побочными продуктами но это побочные процессы.
|
|
|
Гомогенный катализатор находится в той же фазе что и реагенты. Это кислотно–основной катализ, катализ кислотами (акцепторы электронов) и основаниями (доноры электронов) Льюиса, ферментативный катализ.
Скорость химической реакции
Дадим определение основному понятию химической кинетики – скорости химической реакции:
Скорость химической реакции есть число элементарных актов химической реакции, происходящих в единицу времени в единице объема (для гомогенных реакций) или на единице поверхности (для гетерогенных реакций).
Скорость химической реакции есть изменение концентрации реагирующих веществ в единицу времени.
Первое определение является наиболее строгим; из него следует, что скорость химической реакции можно также выражать как изменение во времени любого параметра состояния системы, зависящего от числа частиц какого-либо реагирующего вещества, отнесенное к единице объема или поверхности – электропроводности, оптической плотности, диэлектрической проницаемости и т.д. и т.п. Однако наиболее часто в химии рассматривается зависимость концентрации реагентов от времени. В случае односторонних (необратимых) химических реакций (здесь и далее рассматриваются только односторонние реакции) очевидно, что концентрации исходных веществ во времени постоянно уменьшаются (ΔСисх < 0), а концентрации продуктов реакции увеличиваются (ΔСпрод > 0). Скорость реакции считается положительной, поэтому математически определение средней скорости реакции в интервале времени Δt записывается следующим образом:
 (II.1)
(II.1)
В различных интервалах времени средняя скорость химической реакции имеет разные значения; истинная (мгновенная) скорость реакции определяется как производная от концентрации по времени:
|
|
|
 (II.2)
(II.2)
Графическое изображение зависимости концентрации реагентов от времени есть кинетическая кривая (рисунок 2.1).

Рис. 2.1 Кинетические кривые для исходных веществ (А) и продуктов реакции (В).
Истинную скорость реакции можно определить графически, проведя касательную к кинетической кривой (рис. 2.2); истинная скорость реакции в данный момент времени равна по абсолютной величине тангенсу угла наклона касательной: 
Рис. 2.2 Графическое определение Vист.
 (II.3)
(II.3)
Необходимо отметить, что в том случае, если стехиометрические коэффициенты в уравнении химической реакции неодинаковы, величина скорости реакции будет зависеть от того, изменение концентрации какого реагента определялось. Очевидно, что в реакции
2Н2 + О2 ––> 2Н2О
концентрации водорода, кислорода и воды изменяются в различной степени:
ΔС(Н2) = ΔС(Н2О) = 2 ΔС(О2).
Скорость химической реакции зависит от множества факторов: природы реагирующих веществ, их концентрации, температуры, природы растворителя и т.д.
Кинетическое уравнение химической реакции. Порядок реакции.
Одной из задач, стоящих перед химической кинетикой, является определение состава реакционной смеси (т.е. концентраций всех реагентов) в любой момент времени, для чего необходимо знать зависимость скорости реакции от концентраций. В общем случае, чем больше концентрации реагирующих веществ, тем больше скорость химической реакции. В основе химической кинетики лежит т. н. основной постулат химической кинетики:
Скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ, взятых в некоторых степенях.
Т. е. для реакции
аА + bВ + dD +... ––> еЕ +...
можно записать:
 (II.4)
(II.4)
Коэффициент пропорциональности k есть константа скорости химической реакции. Константа скорости численно равна скорости реакции при концентрациях всех реагирующих веществ, равных 1 моль/л.
Зависимость скорости реакции от концентраций реагирующих веществ определяется экспериментально и называется кинетическим уравнением химической реакции. Очевидно, что для того, чтобы записать кинетическое уравнение, необходимо экспериментально определить величину константы скорости и показателей степени при концентрациях реагирующих веществ. Показатель степени при концентрации каждого из реагирующих веществ в кинетическом уравнении химической реакции (в уравнении (II.4) соответственно x, y и z) есть частный порядок реакции по данному компоненту. Сумма показателей степени в кинетическом уравнении химической реакции (x + y + z) представляет собой общий порядок реакции. Следует подчеркнуть, что порядок реакции определяется только из экспериментальных данных и не связан со стехиометрическими коэффициентами при реагентах в уравнении реакции. Стехиометрическое уравнение реакции представляет собой уравнение материального баланса и никоим образом не может определять характера протекания этой реакции во времени.
|
|
|
В химической кинетике принято классифицировать реакции по величине общего порядка реакции. Рассмотрим зависимость концентрации реагирующих веществ от времени для необратимых (односторонних) реакций нулевого, первого и второго порядков.
Реакции нулевого порядка
Для реакций нулевого порядка кинетическое уравнение имеет следующий вид:
 (II.5)
(II.5)
Скорость реакции нулевого порядка постоянна во времени и не зависит от концентраций реагирующих веществ; это характерно для многих гетерогенных (идущих на поверхности раздела фаз) реакций в том случае, когда скорость диффузии реагентов к поверхности меньше скорости их химического превращения.
Реакции первого порядка
Рассмотрим зависимость от времени концентрации исходного вещества А для случая реакции первого порядка А ––> В. Реакции первого порядка характеризуются кинетическим уравнением вида (II.6). Подставим в него выражение (II.2):
 (II.6)
(II.6)
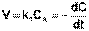 (II.7)
(II.7)
После интегрирования выражения (II.7) получаем:
 (II.8)
(II.8)
Константу интегрирования g определим из начальных условий: в момент времени t = 0 концентрация С равна начальной концентрации Со. Отсюда следует, что g = ln Со. Получаем:
 (II.9)
(II.9)

Рис. 2.3 Зависимость логарифма концентрации от времени для реакций
первого порядка
Т.о., логарифм концентрации для реакции первого порядка линейно зависит от времени (рис. 2.3) и константа скорости численно равна тангенсу угла наклона прямой к оси времени.
 (II.10)
(II.10)
Из уравнения (II.9) легко получить выражение для константы скорости односторонней реакции первого порядка:
 (II.11)
(II.11)
Еще одной кинетической характеристикой реакции является период полупревращения t1/2 – время, за которое концентрация исходного вещества уменьшается вдвое по сравнению с исходной. Выразим t1/2 для реакции первого порядка, учитывая, что С = ½Со:
|
|
|
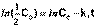 (II.12)
(II.12)
Отсюда
 (II.13)
(II.13)
Как видно из полученного выражения, период полупревращения реакции первого порядка не зависит от начальной концентрации исходного вещества.
Реакции второго порядка
Для реакций второго порядка кинетическое уравнение имеет следующий вид:
 (II.14)
(II.14)
либо
 (II.15)
(II.15)
Рассмотрим простейший случай, когда кинетическое уравнение имеет вид (II.14) или, что то же самое, в уравнении вида (II.15) концентрации исходных веществ одинаковы; уравнение (II.14) в этом случае можно переписать следующим образом:
 (II.16)
(II.16)
После разделения переменных и интегрирования получаем:
 (II.17)
(II.17)
Постоянную интегрирования g, как и в предыдущем случае, определим из начальных условий. Получим:
 (II.18)
(II.18)
Т.о., для реакций второго порядка, имеющих кинетическое уравнение вида (II.14), характерна линейная зависимость обратной концентрации от времени (рис. 2.4) и константа скорости равна тангенсу угла наклона прямой к оси времени:
 (II.19)
(II.19)
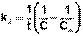 (II.20)
(II.20)

Рис. 2.4 Зависимость обратной концентрации от времени для реакций
второго порядка
Если начальные концентрации реагирующих веществ Cо,А и Cо,В различны, то константу скорости реакции находят интегрированием уравнения (II.21), в котором CА и CВ – концентрации реагирующих веществ в момент времени t от начала реакции:
 (II.21)
(II.21)
В этом случае для константы скорости получаем выражение
 (II.22)
(II.22)
Порядок химической реакции есть формально-кинетическое понятие, физический смысл которого для элементарных (одностадийных) реакций заключается в следующем: порядок реакции равен числу одновременно изменяющихся концентраций. В случае элементарных реакций порядок реакции может быть равен сумме коэффициентов в стехиометрическом уравнении реакции; однако в общем случае порядок реакции определяется только из экспериментальных данных и зависит от условий проведения реакции. Рассмотрим в качестве примера элементарную реакцию гидролиза этилового эфира уксусной кислоты (этилацетата), кинетика которой изучается в лабораторном практикуме по физической химии:
СН3СООС2Н5 + Н2О ––> СН3СООН + С2Н5ОН
Если проводить эту реакцию при близких концентрациях этилацетата и воды, то общий порядок реакции равен двум и кинетическое уравнение имеет следующий вид:
 (II.23)
(II.23)
При проведении этой же реакции в условиях большого избытка одного из реагентов (воды или этилацетата) концентрация вещества, находящегося в избытке, практически не изменяется и может быть включена в константу скорости; кинетическое уравнение для двух возможных случаев принимает следующий вид:
1) Избыток воды:
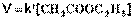 (II.24)
(II.24)
 (II.25)
(II.25)
2) Избыток этилацетата:
 (II.26)
(II.26)
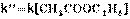 (II.27)
(II.27)
В этих случаях мы имеем дело с т.н. реакцией псевдопервого порядка. Проведение реакции при большом избытке одного из исходных веществ используется для определения частных порядков реакции.
КАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ
Скорость химической реакции при данной температуре определяется скоростью образования активированного комплекса, которая, в свою очередь, зависит от величины энергии активации. Во многих химических реакциях в структуру активированного комплекса могут входить вещества, стехиометрически не являющиеся реагентами; очевидно, что в этом случае изменяется и величина энергии активации процесса. В случае наличия нескольких переходных состояний реакция будет идти в основном по пути с наименьшим активационным барьером.
Катализ – явление изменения скорости химической реакции в присутствии веществ, состояние и количество которых после реакции остаются неизменными.
Различают положительный и отрицательный катализ (соответственно увеличение и уменьшение скорости реакции), хотя часто под термином "катализ" подразумевают только положительный катализ; отрицательный катализ называют ингибированием.
Вещество, входящее в структуру активированного комплекса, но стехиометрически не являющееся реагентом, называется катализатором. Для всех катализаторов характерны такие общие свойства, как специфичность и селективность действия.
Специфичность катализатора заключается в его способности ускорять только одну реакцию или группу однотипных реакций и не влиять на скорость других реакций. Так, например, многие переходные металлы (платина, медь, никель, железо и т.д.) являются катализаторами для процессов гидрирования; оксид алюминия катализирует реакции гидратации и т.д.
Селективность катализатора – способность ускорять одну из возможных при данных условиях параллельных реакций. Благодаря этому можно, применяя различные катализаторы, из одних и тех же исходных веществ получать различные продукты:
| [Cu]: СО + Н2 ––> СН3ОН | [Al2О3]: С2Н5ОН ––> С2Н4 + Н2О |
| [Ni]: СО + Н2 ––> СН4 + Н2О | [Cu]: С2Н5ОН ––> СН3СНО + Н2 |
Причиной увеличения скорости реакции при положительном катализе является уменьшение энергии активации при протекании реакции через активированный комплекс с участием катализатора (рис. 2.8).
Поскольку, согласно уравнению Аррениуса, константа скорости химической реакции находится в экспоненциальной зависимости от величины энергии активации, уменьшение последней вызывает значительное увеличение константы скорости. Действительно, если предположить, что предэкспоненциальные множители в уравнении Аррениуса (II.32) для каталитической и некаталитической реакций близки, то для отношения констант скорости можно записать:
 (II.44)
(II.44)
Если ΔEA = –50 кДж/моль, то отношение констант скоростей составит 2,7·106 раз (действительно, на практике такое уменьшение EA увеличивает скорость реакции приблизительно в 105 раз).
Необходимо отметить, что наличие катализатора не влияет на величину изменения термодинамического потенциала в результате процесса и, следовательно, никакой катализатор не может сделать возможным самопроизвольное протекание термодинамически невозможного процесса (процесса, ΔG (ΔF) которого больше нуля). Катализатор не изменяет величину константы равновесия для обратимых реакций; влияние катализатора в этом случае заключается только в ускорении достижения равновесного состояния.
В зависимости от фазового состояния реагентов и катализатора различают гомогенный и гетерогенный катализ.

Рис. 2.8 Энергетическая диаграмма химической реакции без катализатора (1)
и в присутствии катализатора (2).
2.3.1 Гомогенный катализ.
Гомогенный катализ – каталитические реакции, в которых реагенты и катализатор находятся в одной фазе. В случае гомогенно-каталитических процессов катализатор образует с реагентами промежуточные реакционноспособные продукты. Рассмотрим некоторую реакцию
А + В ––> С
В присутствии катализатора осуществляются две быстро протекающие стадии, в результате которых образуются частицы промежуточного соединения АК и затем (через активированный комплекс АВК#) конечный продукт реакции с регенерацией катализатора:
А + К ––> АК
АК + В ––> С + К
Примером такого процесса может служить реакция разложения ацетальдегида, энергия активации которой EA = 190 кДж/моль:
СН3СНО ––> СН4 + СО
В присутствии паров йода этот процесс протекает в две стадии:
СН3СНО + I2 ––> СН3I + НI + СО
СН3I + НI ––> СН4 + I2
Уменьшение энергии активации этой реакции в присутствии катализатора составляет 54 кДж/моль; константа скорости реакции при этом увеличивается приблизительно в 105 раз. Наиболее распространенным типом гомогенного катализа является кислотный катализ, при котором в роли катализатора выступают ионы водорода Н+.
Автокатализ.
Автокатализ – процесс каталитического ускорения химической реакции одним из её продуктов. В качестве примера можно привести катализируемую ионами водорода реакцию гидролиза сложных эфиров. Образующаяся при гидролизе кислота диссоциирует с образованием протонов, которые ускоряют реакцию гидролиза. Особенность автокаталитической реакции состоит в том, что данная реакция протекает с постоянным возрастанием концентрации катализатора. Поэтому в начальный период реакции скорость её возрастает, а на последующих стадиях в результате убыли концентрации реагентов скорость начинает уменьшаться; кинетическая кривая продукта автокаталитической реакции имеет характерный S-образный вид (рис. 2.9).

Рис. 2.9 Кинетическая кривая продукта автокаталитической реакции
Гетерогенный катализ.
Гетерогенный катализ – каталитические реакции, идущие на поверхности раздела фаз, образуемых катализатором и реагирующими веществами. Механизм гетерогенно-каталитических процессов значительно более сложен, чем в случае гомогенного катализа. В каждой гетерогенно-каталитической реакции можно выделить как минимум шесть стадий:
1. Диффузия исходных веществ к поверхности катализатора.
2. Адсорбция исходных веществ на поверхности с образованием некоторого промежуточного соединения:
А + В + К ––> АВК
3. Активация адсорбированного состояния (необходимая для этого энергия есть истинная энергия активации процесса):
АВК ––> АВК#
4. Распад активированного комплекса с образованием адсорбированных продуктов реакции:
АВК# ––> СDК
5. Десорбция продуктов реакции с поверхности катализатора.
СDК ––> С + D + К
6. Диффузия продуктов реакции от поверхности катализатора.
Специфической особенностью гетерокаталитических процессов является способность катализатора к промотированию и отравлению.
Промотирование – увеличение активности катализатора в присутствии веществ, которые сами не являются катализаторами данного процесса (промоторов). Например, для катализируемой металлическим никелем реакции
СО + Н2 ––> СН4 + Н2О
введение в никелевый катализатор небольшой примеси церия приводит к резкому возрастанию активности катализатора.
Отравление – резкое снижение активности катализатора в присутствии некоторых веществ (т. н. каталитических ядов). Например, для реакции синтеза аммиака (катализатор – губчатое железо), присутствие в реакционной смеси соединений кислорода или серы вызывает резкое снижение активности железного катализатора; в то же время способность катализатора адсорбировать исходные вещества снижается очень незначительно.
Для объяснения этих особенностей гетерогенно-каталитических процессов Г. Тэйлором было высказано следующее предположение: каталитически активной является не вся поверхность катализатора, а лишь некоторые её участки – т.н. активные центры, которыми могут являться различные дефекты кристаллической структуры катализатора (например, выступы либо впадины на поверхности катализатора). В настоящее время нет единой теории гетерогенного катализа. Для металлических катализаторов была разработана теория мультиплетов. Основные положения мультиплетной теории состоят в следующем:
1. Активный центр катализатора представляет собой совокупность определенного числа адсорбционных центров, расположенных на поверхности катализатора в геометрическом соответствии со строением молекулы, претерпевающей превращение.
2. При адсорбции реагирующих молекул на активном центре образуется мультиплетный комплекс, в результате чего происходит перераспределение связей, приводящее к образованию продуктов реакции.
Теорию мультиплетов называют иногда теорией геометрического подобия активного центра и реагирующих молекул. Для различных реакций число адсорбционных центров (каждый из которых отождествляется с атомом металла) в активном центре различно – 2, 3, 4 и т.д. Подобные активные центры называются соответственно дублет, триплет, квадруплет и т.д. (в общем случае мультиплет, чему и обязана теория своим названием).
Например, согласно теории мультиплетов, дегидрирование предельных одноатомных спиртов происходит на дублете, а дегидрирование циклогексана – на секстете (рис. 2.10 – 2.11); теория мультиплетов позволила связать каталитическую активность металлов с величиной их атомного радиуса.
 Рис. 2.10 Дегидрирование спиртов на дублете
Рис. 2.10 Дегидрирование спиртов на дублете
 Рис. 2.11 Дегидрирование циклогексана на секстете
Рис. 2.11 Дегидрирование циклогексана на секстете
Ферментативный катализ.
Ферментативный катализ – каталитические реакции, протекающие с участием ферментов – биологических катализаторов белковой природы. Ферментативный катализ имеет две характерные особенности:
1. Высокая активность, на несколько порядков превышающая активность неорганических катализаторов, что объясняется очень значительным снижением энергии активации процесса ферментами. Так, константа скорости реакции разложения перекиси водорода, катализируемой ионами Fе2+, составляет 56 с-1; константа скорости этой же реакции, катализируемой ферментом каталазой, равна 3.5·107, т.е. реакция в присутствии фермента протекает в миллион раз быстрее (энергии активации процессов составляют соответственно 42 и 7.1 кДж/моль). Константы скорости гидролиза мочевины в присутствии кислоты и уреазы различаются на тринадцать порядков, составляя 7.4·10-7 и 5·106 с-1 (величина энергии активации составляет соответственно 103 и 28 кДж/моль).
2. Высокая специфичность. Например, амилаза катализирует процесс расщепления крахмала, представляющего собой цепь одинаковых глюкозных звеньев, но не катализирует гидролиз сахарозы, молекула которой составлена из глюкозного и фруктозного фрагментов.
Согласно общепринятым представлениям о механизме ферментативного катализа, субстрат S и фермент F находятся в равновесии с очень быстро образующимся фермент-субстратным комплексом FS, который сравнительно медленно распадается на продукт реакции P с выделением свободного фермента; т.о., стадия распада фермент-субстратного комплекса на продукты реакции является скоростьопределяющей (лимитирующей).
F + S <––> FS ––> F + P
Исследование зависимости скорости ферментативной реакции от концентрации субстрата при неизменной концентрации фермента показали, что с увеличением концентрации субстрата скорость реакции сначала увеличивается, а затем перестает изменяться (рис. 2.12) и зависимость скорости реакции от концентрации субстрата описывается следующим уравнением:
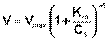 (II.45)
(II.45)
Здесь Кm – константа Михаэлиса, численно равная концентрации субстрата при V = ½Vmax. Константа Михаэлиса служит мерой сродства между субстратом и ферментом: чем меньше Кm, тем больше их способность к образованию фермент-субстратного комплекса.
Характерной особенностью действия ферментов является также высокая чувствительность активности ферментов к внешним условиям – рН среды и температуре. Ферменты активны лишь в достаточно узком интервале рН и температуры, причем для ферментов характерно наличие в этом интервале максимума активности при некотором оптимальном значении рН или температуры; по обе стороны от этого значения активность ферментов быстро снижается.

|
|
|
