 |


Дифференциация популяций по грузу наследственных болезней 21 глава
|
|
|
|
Доминантное наследование аномалии в виде короткопалости (брахидактилии) у человека впервые установил американский антрополог Farabee, описавший в своей докторской диссертации семью с этой аномалией, в которой в последующие годы было установлено 43 индивида с брахидактилией в 7 поколениях Интересно отметить что у 43 детей имелась брахидактилия, а у 45 были нормальные пальцы, что соответствует теоретически ожидаемому распределению 1 1 У лиц с нормальными пальцами все дети и внуки также имели нормальные пальцы и не отмечалось «проскакивающего» поколения
Каждый ген от гетерозигот в среднем передается половине детей Из каждой пары генов одного из родителей ребенок всегда получает тот или иной ген Различия в «типах наследования» проявляют не гены, а фенотипические признаки Некоторые доминантные гены, например, пельгеровская аномалия лейкоцитов, не приводят к заметным тяжелым патологическим дефектам и могут передаваться через многие поколения Поэтому их чаще обнаруживают у одного из родите- лей-носителя признака и в среднем у 50% его сибсов и детей Патологические доминантные гены встречаются намного реже, чем рецессивные, и поэтому браки между двумя носителями патологических доминантных генов крайне редки, но только от таких браков и могут произойти лица, которые в гетерозиготном состоянии обусловливают развитие заболевания Подобное наблюдение описано в литературе, когда в двух браках между двумя родителями с ахондроппазией родилось по одному ребенку с чрезвычайно тяжелыми изменениями трубчатых костей, ребер, пальцев, тел позвонков и таза Это объясняется патологическим дефектом белка у гомозигот, а у гетерозигот 50% его составляет нормальный белок
|
|
|
Следует отметить, что некоторые ауто- сомно-доминантные болезни (хорея Ген- тингтона, поликистозная болезнь почек взрослых, миотоническая дистрофия и др) в детском возрасте могут не обнару живаться, а взрослые носители заболеваний могут быть внешне здоровыми и только в определенный период жизни (мини мальный критический период проявления) у них появляются первые симптомы болезни Заболевания с поздним началом не редко передаются через многие поколения Развитие заболевания определяется также пенетрантностью (процент проявле ния болезни среди носителей патологического гена) и экспрессивностью (степень выраженности фенотипа) гена Доминант ные гены, распространению которых препятствуют высокая летальность, низкие физические, умственные способности и низкая плодовитость, редко передаются детям Однако пополнение этого класса заболеваний обеспечивают новые мута ции, которые определяют спорадический характер болезней, пока пораженные ли ца не имеют детей
Частота возникновения разнообразных мутаций генов у человека, лежащих в основе аутосомно доминантных, аутосомно- рецессивных и Х-сцепленных форм, представлена в табл 4 2 0 1
При нейрофиброматозе и туберозном склерозе около половины случаев являются спорадическими, и примерно наполовину от нормы снижена репродуктивная способность больных Напротив, нетяжелые доминантные аномалии (пельгеровская аномалия лейкоцитов, эллиптоцитоз, че- репно-ключичный дизостоз и др) часто об наруживают при анализе больших родо словных
Таким образом, наиболее характерны ми чертами аутосомно-доминантных признаков и болезней являются следующие
1 Для доминантного гена характерны полная или высокая проявляемость в гете розиготном состоянии, заболевание ветре чается почти в каждом поколении Соотношение здоровых и больных сибсов в данном поколении около 1 1
|
|
|
2 Пенетрантность доминантных генов довольно высока, а доминантным заболе ваниям свойственна варьирующая экспрессивность
3 Клинические проявления заболева ний отличаются не только значительной межсемейной, но и внутрисемейной вариа бельностью
4 Манифестация ряда доминантных болезней может быть поздней или носить варьирующий характер
5 Гомозиготное состояние для доминантно наследуемых болезней встречается крайне редко, а клинические проявления доминантных болезней в гомозиготном состоянии отличаются особой тяжестью
6 При аутосомно-доминатных заболеваниях отсутствует преимущественное поражение того или иного пола
7 Заболевание часто носит спорадический характер
Несмотря на то, что многие моногенные болезни обязаны своим происхождением новым мутациям, вопрос об одинаковой или разной частоте возникновения доминантных и рецессивных мутаций остается недостаточно исследованным Согласно современным представлениям, частота мутаций у человека равна 1-2 на 100 ООО гамет и реже При ряде заболеваний (синдром Марфана, оссифицирующий миозит, ахондроплазия, синдром Апера и др) от мечена зависимость частоты мутаций от возраста отцов Однако частота мутаций зависит от многих факторов от физиологического состояния организма, возраста, генотипа, интенсивности мутагенных факторов внешней среды и др Корреляцион ные связи факторов-мутагенов и уровня мутационного процесса не всегда можно установить Результаты проведенных попу ляционных исследований, выполненных в Российской Федерации [1, 2], свидетельствуют о том, что отягощенность моногеннь)- ми формами наследственной патологии довольно высокая (табл 4 2 0 2)
Знание общей характеристики моногенных болезней, передающихся по аутосом-
| Таблица 420 1 Частота возникновения разиввбраз- | |
| иых мутаций у человека (обобщенные данные ли | |
| тературы) | |
| Группы заболеваний Количество | |
| мутаций | |
| на 1 млн. | |
| гамет | |
| 1. Аутосомно-доминантные болезни и признаки | |
| Поликистоз почек | 65-120 |
| Нейрофиброматоз Реклингаузена | 44-100 |
| Множественный полипоз | |
| толстого кишечника | 10-50 |
| Болезнь Марфана | 4,2-5,8 |
| Пельгеровская аномалия лейкоцитов | 9-27 |
| Ретинобластома | 3-12 |
| Туберозный склероз | 6-10,5 |
| Хорея Гентингтона | 1-10 |
| Несовершенный остеогенез | 7-13 |
| Множественная хондродисплазия | 6,3-9,1 |
| Акроцефалосиндактилия | 3-4 |
| Ахондроплазия | 5,1-13 |
| Аниридия | 2-3 |
| Микрофтальмия | |
| Синдром Гиппеля-Линдау | 0,18 |
| Лице-лопаточно-плечевая мышечная | |
| дистрофия Ландузи-Дежерина | 6-11 |
| 2. Аутосомно-рецессивные заболевания | |
| Микроцефалия | |
| Амавротичвская идиотия | |
| Буллезный элидермолиз | |
| Врожденный ихтиоз | |
| 3. Х-сцепленные рецессивные болезни | |
| Мышечная дистрофия Дюшенна/Бекера | 43-105 |
| Гемофилия А | 37-52 |
| Гемофилия В | 2-3 |
| Ихтиоз | |
| Глазо-лице-пальцевой синдром |
но-доминантному типу, позволяет педиатру рано заподозрить наследственную природу заболевания, поставить правильный диагноз и дать квалифицированный медико-генетический прогноз.
|
|
|
Перечень аутосомно-доминантных наследственных заболеваний довольно велик. В настоящем разделе нашли отраже
Табяица 4202 Число новых случаев менделирующих болезней в Российской Федерации (за 1 год иа 1 млн. населеиия [2]
| Типы наследственных болезней | Число |
| новых | |
| случаев | |
| Аутосомно-доминантные | 15,2 |
| Аутосомно-рецвссивные | 13,2 |
| Х-сцепленные | 3,6 |
| Все менделирующие болезни | 32,0 |
ние наиболее значимые для педиатрической практики нозологические формы патологии.
4.2.1. Синдром Марфана
Синдром Марфана относится к моногенным болезням соединительной ткани - группе различных по происхождению нозологических форм, которые объединяют наследственные нарушения обмена соединительной ткани. Большая часть этой патологии обусловлена нарушением ферментных систем, контролирующих синтез структурных белков. Почти все эти болезни приводят к тяжелым инвалидизирующим расстройствам.
Синдром Марфана впервые описан Вильямсом в 1876 г. Свое название заболевание получило от французского педиатра Марфана, наблюдавшего девочку с характерным симптомокомплексом болезни 20 лет спустя.
Частота синдрома Марфана в общей популяции составляет 4: 100 ООО [3].
Гэнетические данные и патогенез. Заболевание наследуется по аутосомно-до- минантному типу с высокой пенетрантно- стью мутантного гена [3]. Однако в 1992 г. мексиканские исследователи [4] на основании наблюдения трех поколений одной семьи, в которой пять из шести сибсов имели типичные признаки синдрома Марфана, высказали предположение о возможности аутосомно-рецессивного наследования патологии. Трое из этих детей (одна девочка и два мальчика) погибли от разрыва аневризмы аорты в возрасте 9, 12 и 14 лет. На основании отсутствия у больных умственной отсталости и метаболических нарушений была исключена го- моцистинурия. Тщательное обследование ближайших родственников пробандов (родителей сибсов, их дедов, бабушек и дяди по отцовской линии) не выявили у них признаков синдрома Марфана. Все это позволило авторам отвергнуть аутосомно-доми- нантное наследование заболевания, появление свежих мутаций и трактовать данный случай как пример возможного ауто- сомно-рецессивного наследования синдрома Марфана.
|
|
|
Мутантный ген FBN1 локализован на длинном плече хромосомы 15, в локусе q 12,1. Этот ген кодирует синтез гликопроте- ина внеклеточного матрикса фибриллина- 1 (5, 6]. Ген FBN1 имеет размер более 200 т.п.н., состоит из 65 экзонов, что крайне затрудняет прямой анализ мутаций в этом гене. По этой же причине до сих пор остаются бездоказательными генотип-феноти- пические корреляции [5]. В настоящее время для диагностики семейных случаев синдрома Марфана, как правило, используется непрямой метод, основанный на применении высокополиморфных тесно сцепленных с геном FBN1 микросателлит- ных ДНК-маркеров. Разработана панель из девяти ДНК-маркеров и доказана ее высокая информативность (практически для всех 100% семей с синдромом Марфана) [6]. Анализ спорадических случаев заболевания (составляющий 25-30% от всей патологии), напротив, базируется на методе прямой диагностики мутаций в гене FBN1. Преимущества прямой диагностики синдрома Марфана очевидны и заключаются в выявлении как точковых мутаций, так и мутаций сплайсинга. С помощью этого метода возможно также пренаталь- ное установление диагноза [7]. В основе болезни лежит недостаточность синтеза фибриллина-1 [3].
Клинические проявления. Клиническая картина заболевания характеризуется поражением многих жизненно важных органов и систем: опорно-двигательного аппарата, сердечно-сосудистой системы, органов дыхания и зрения, ЦНС. Так, среди конституциональных особенностей и нарушений скелета наиболее часто встречаются долихопласти- ческий (астенический) морфотип, высокий рост (как правило, выше 97 центиля) при выраженном дефиците массы тела (обычно ниже третьего центиля), арахно- дактилия («паучьи» пальцы) кистей и стоп, кифосколиоз, воронкообразная или килевидная деформации грудной клетки, плоскостопие, узкий лицевой скелет, «готическое» небо [6, 7). Типичны также антимонголоидный разрез глаз, «крупный» нос, большие низкорасположенные ушные раковины, «птичье» выражение лица (рис. 4.2 1.1. и 4.2.1.2.).
|
|
|
В периоде новорожденности из перечисленных особенностей скелета, как правило, выявляются только долихопластиче- ский морфотип и арахнодактилия. Остальные симптомы формируются в более поздние периоды постнатального развития (обычно в течение первых семи лет жизни ребенка) [9].
Поражение сердца и сосудов - один из кардинальных признаков синдрома Мар-
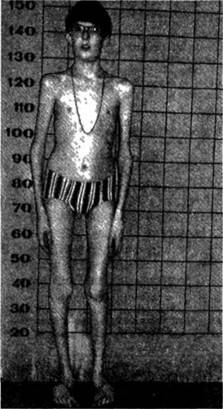
Рис 4 211 Ребенок 12 лет с еиидремем Марфана. Делихопластический мерфотип, узкий лицевем скелет, «птичье» лицо.
фана. Наиболее типичны среди них - пролапс митрального клапана и аневризма аорты. Сердечно-сосудистые нарушения регистрируются уже на первом-втором годах жизни ребенка, при этом отмечается постепенное увеличение диаметра аорты, достигающее критических размеров (до б см и более), чаще в возрасте от 16 до 45 лет. Грозным осложнением аневризмы аорты является расслоение ее стенок, которое может быстро прогрессировать, захватывая всю длину аорты и отходящие от нее сосуды. Такие осложнения, как правило, заканчиваются летально.
Бронхолегочная система также вовлекается в патологический процесс при синдроме Марфана. Предпосылкой для этого являются механическое сдавление дыхательных путей при деформациях грудной клетки и изменения соединительно-тканных структур легочной ткани. Нарушения органов дыхания в виде спонтанного пневмоторакса, легочной эмфиземы, инфаркта легкого встречаются с частотой от 10 до 75% [3, 9]. Наряду с этим, имеются сведения о врожденном недоразвитии одной из долей легкого, поликистозе легких, врожденной буллезной эмфиземе, двусторонних бронхоэктазах [3, 10].
К наиболее типичной патологии органа зрения при синдроме Марфана относится вывих и подвывих хрусталика (вследствие слабости цинновой связки). Как правило, эта патология сочетается с миопией или гиперметропией высокой степени [3, 10, 11]. Подвывих хрусталиков диагностируется обычно на 1-5 годах жизни, а иногда даже в 7 лет при оформлении ребенка в школу. Реже встречаются вторичная глаукома, катаракта, отслойка сетчатки. Эти изменения чаще выявляются у больных более старшего возраста - 15-18-40 лет [11].
IQ (коэффициент интеллектуального развития) у большинства детей с синдромом Марфана обычно соответствует норме - 85-115 единиц. Встречаются лица с очень высоким интеллектом, у которых

Рис 4212 Девочка 10 лет с синдромом Марфана. Длинные тонкие конечнести, высокий рост, диссоциация маооороотовых параметров, кифосколиоз.
Ю превышает верхнюю границу нормы - 115 ед. Так, установлено, что ряд всемирно известных людей страдали синдромом Марфана, среди них следует упомянуть президента США А.Линкольна и великого скрипача Паганини [8]. Однако может иметь место определенное своеобразие психических процессов, которое проявляется в неравномерной интеллектуальной деятельности, а также в личностных особенностях больных (раздражительности, плаксивости, завышенной самооценке) [9].
Для всех детей с синдромом Марфана характерна низкая переносимость физической нагрузки, которая нередко сопровождается болями в мышцах. Возможны также периодические приступы мигрене- подобной головной боли, возникающей, как правило, на фоне или после эмоционально-физических нагрузок [12]. Эти признаки заболевания в сочетании со слабо стью, гипотонией и гипоплазией мышеч ной ткани, а также нарушениями показа телей физического развития служат сви детельством изменения функции митохондрий (нарушением процессов клеточной биоэнергетики)
Диагностика синдрома Марфана бази руется на генеалогических данных (составление и анализ родословных) и анализе морфофенотипа пробанда, который вклю чает изучение физического, нервно-психического развития детей и состояния различных органов и систем организма
Оценка физического развития больных проводится с помощью перцентильных шкал Стюарта О пропорциональности или гармоничности отдельных частей тела судят путем использования индекса Дю Ранта-Лайнера [15], который вычисляется по формуле А/В х 100, где А - отношение фа ктической массы тела к 50 перцентилю массы, соответствующей росту больного, а В - отношение фактической длины тела к 50 перцентилю роста соответствующего возраста Индекс количественно отражает вариации физического развития При этом показатель 89 и ниже соответствует высокому росту при дефиците массы тела, показатели - 110-119 - избыточной массе тела, свыше 120 - ожирению [8] Для детей с синдромом Марфана индекс Дю Ранта-Лайнера, как правило, составляет 51-81 [13]
По резолюции совещания, посвященного синдрому Марфана [15], для поста новки диагноза необходимо наличие ми нимум одного из пяти основных симптомов заболевания (вывих хрусталиков, аневризма аорты, арахнодактилия, деформация грудины, кифосколиоз) и двух дополнительных (миопия, пролапс мит рапьного клапана, умеренная гиперпо движность суставов высокий рост, плоскостопие, стрии пневмоторакс) Установлено что в 90% всех случаев синдрома Марфана трудностей в постановке пра вильного диагноза, как правило, не возникает Однако в 10% - диагностика затруднена В подобных ситуациях особенно необходимо чрезвычайно тщательное обследование максимально большего числа родственников больного Программа обследования для таких семей должна обя зательно включать консультативные ос мотры окулиста, кардиолога и проведе ниє эхокардиографии
В диагностике синдрома Марфана ши роко используются также результаты рентгено-функциональных методов ис следования Так, для оценки арахнодак- тилии используют показатели метакар- пального индекса (отношение длины к ширине второй-пятой метакарпальных костей), вычисляемого по рентгенограмме правой кисти У больных с синдромом Марфана наблюдается увеличение этого показателя до 8,0-11,0 при норме 6,4-7,9 [10 13]
Характер и степень тяжести сердечно сосудистой патологии оценивают по дан ным эхокардиографии, ЭКГ, холтеровско му мониторированию Анализ состояния бронхолегочной системы проводят по ре зультатам исследования функции внешне го дыхания У подавляющего числа детей с синдромом Марфана регистрируются изменения этих показателей проявляющиеся в нарушении механики дыхания, вздутии легочной ткани, неравномерности распределения в легких вдыхаемого воздуха гиперкапнии [9]
У детей с синдромом Марфана обнару живается снижение репарационной способности ДНК-лимфоцитов, что необходи мо принимать во внимание при рентгено логическом обследовании выборе профессии и места жительства больных [11 ]
При синдроме Марфана имеет место увеличение (в два раза и более) выведе ния с мочой гпикозаминогликанов и их фракций, при этом особенно резко воз растает почечная экскреция хондроитин-4 6 сульфатов и в меньшей степени - гиалу роновой кислоты и гепаран-сульфата Наиболее высокая концентрация в моче хон дроитин-4-6-сульфатов отмечается у детей, в клинической картине которых преобладают грубые скелетные нарушения. В моче обследованных больных с синдромом Марфана определяется также повышенное содержание (в два раза и более) иминокислоты оксипролина. При этом выявляется четкая зависимость показателей экскреции оксипролина от тяжести заболевания и возраста больных: наибольшие значения отмечаются у детей с выраженной клинической симптоматикой и в возрасте от 8 до 18 лет. При изучении выделения с мочой оксилизилгликозидов у 90% детей с синдромом Марфана обнаруживают их превышение и изменение соотношения дисахаридной и моносахаридной фракций [11].
О нарушении процессов клеточной биоэнергетики у больных с синдромом Марфана свидетельствует ряд биохимических параметров: умеренное повышение уровня молочной и пировиноградной кислот в сыворотке крови, оцениваемое на фоне стандартного глюкозотолерант- ного теста (1,75 г/кг массы тела); снижение содержания макроэргов в крови (АТФ, АДФ, АМФ) и изменения процессов перекисного окисления липидов, характеризующиеся увеличением показателей малонового диальдегида, гидроперекисей и снижением антиокислительной активности плазмы.
Морфологическими критериями мито- хондриальной недостаточности являются: наличие «рваных красных волокон» в био- птате скелетной мышцы в количестве от 20% до 40% (при норме до 5%), субсарко- леммальные скопления гликогена, липидов и кальция при сохранной активности митохондриальных ферментов (цитохро- моксидазы, сукцинатдегидрогеназы) [13].
| Таблица 4211 Дифференциальная диагностика синдрома Марфана с феиотипичвски сходными заболеваниями | |||||
| Признаки заболевания | Синдромы | ||||
| Билса Стиплера Вейла-Марчезани Марфана гомоцистинурия | |||||
| Аутосомно-доминантный тип наследования | + | + | - | + | - |
| Аутосомно-рецессивный тип наследования | - | - | + | - | + |
| Астеническое телосложение | + | + | - | + | + |
| Гилерстеническое телосложение | - | - | + | - | - |
| Арахнодактилия | + | + | - | + | + |
| Деформация грудной клетки | +/- | - | - | + /- | + /- |
| Кифосколиоз | +/- | - | + | + | |
| Врожденные контрактуры суставов | + | - | - | - | - |
| Артрозоартриты | - | + | - | - | - |
| «Мятое» ухо | + | - | - | - | - |
| ВрЬжденные пороки сердца | - | - | + /- | +/- | - |
| Пролапс митрального клапана | +/- | +/- | + /- | + | + /- |
| Аневризма аорты | - | - | - | + | - |
| Тромбоэмболии | - | - | - | - | + |
| Подвывих хрусталиков | - | - | +/- | + | + |
| Микросферофокия | - | - | + | - | - |
| Миопия высокой степени | - | + | +/- | + | + /- |
| Снижение интеллекта | - | - | - | - | + |
| Параличи и парезы | - | - | - | - | + |
| Повышение уровня метионина, появление | |||||
| гомоцистина в сыворотке крови и моче | - | - | - | - | + |
Диффренциальная диагностика. Основные дифференциально диагностические признаки синдрома Марфана и фе- нотипически сходных заболеваний суммированы в табл. 4.2.1.1. Синдром Марфана следует также дифференцировать с «мар- фаноидными» типами синдрома Элер- са-Данлоса. Однако, по данным исследователей [15], поражение аорты при синдроме Элерса-Данлоса встречается значительно реже и регистрируется лишь у 3,7% больных. Наряду с этим, при синдроме Марфана, как правило, отсутствуют такие типичные для синдрома Элер- са-Данлоса изменения кожи, как «псевдоопухоли» и «папиросные» рубцы Обра зование стрий у лиц с синдромом Марфана также считается дополнительным диагностическим критерием для разграничения этих заболеваний[15]
«Марфаноподобный» фенотип имеют больные с множественной эндокринной не- оплазией IIB типа (МЭН ИВ) [9] Однако наличие подслизистых неврином, огрубляющих черты лица больных, и отсутствие поражения сердца и крупных сосудов (аорты) позволяют исключить синдром Марфана у этих детей
Аномалии половых хромосом - синдромы Клайнфельтера (47.XXY) и 47.XYY также необходимо дифференцировать с синдромом Марфана Более грубое снижение интеллекта, отсутствие патологии глаз и сердечнососудистой системы, наряду с результатами цитогенетических исследований, позволяют поставить правильный диагноз
Лечение. Детям с синдромом Марфана показана комплексная терапия, включающая широкий спектр лекарственных препаратов средства, влияющие на сердечнососудистую систему, стимуляторы ЦНС, энерготропные препараты и антиоксидан- ты по схеме «Комплекс терапевтических воздействий, применяемый для лечения больных с синдромом Марфана»
Бета-адреноблокаторы - обзидан, ате- нолол - 10 мг/сут, длительность 6-12 и более мес
Энерготропные и антиоксидантные препараты
• рибоксин - 1 табл (0,2) 2 раза в день в течение 1 мес, 3 курса в год,
• витамины В,, В2 - 10 мг/сут, 10 дней ежемесячно,
• аскорбиновая кислота до 500 мг/сут, в течение 1 мес, 3-4 курса в год,
• токоферол (вит Е) до 100 мг/сут, в течение 3-4-месячных курсов в год,
• элькар - 200-400 мг/сут, 3 мес,
2- 3 курса в год,
• димефосфон - 30 мг/кг - 1 мес,
3- 4 курса в год,
• коэнзим Q-10 - 30 мг 2-3 раза в сут, З мес, 2-3 курса в год,
• лимонтар - 5 мг/кг/сут, 10 дней, 4 курса в год,
• ноотропные препараты пирацетам - 200-400 мг 2 раза в сут в течение 2 мес, 3 курса в год
Наряду с медикаментозными средствами, детям с синдромом Марфана необходим также комплекс дополнительных лечебных воздействий, включающий маг- нитотерапию на суставы (курс 10 сеансов, 3 курса в год), электросон (курс 10 сеансов - дважды в год), лечебную физкультуру с преимущественным воздействием на опорно-двигательный аппарат (курс 14 дней, 4 курса в год), санатории для больных с нарушениями функций костей и суставов или сердечно-сосудистой системы (курс лечения 24 дня - 1 раз в год)
По показаниям, проводится оперативное лечение торакопластика, аневризм- эктомия, пластика аорты, экстракция хрусталика, тонзиллэктомия и аденото- мия Осуществляется также регулярная санация (не менее двух раз в год) хронических очагов инфекции полости рта и зубов
Под воздействием комплексной терапии у 78-80% детей с синдромом Марфана отмечаются улучшение или стабилизация основного патологического процесса Клиническими критериями эффективности проводимого лечения служат повышение толерантности к физическим нагрузкам, нарастание мышечной силы, стабилизация диаметра аорты (по данным эхокардиографии) и тенденция к нормализации показателей функции внешнего дыхания, улучшение мелкой моторики, повышение эмоционального тонуса, увеличение объема про извольной памяти и концентрации внима ния, повышение школьной успеваемости
Положительная биохимическая динамика проявляется в снижении уровня молочной и пировиноградной кислот, уменьшении содержания малонового диальдегида и гидроперекисей, увеличении антиокислительной активности плазмы и суммарного содержания макроэргов.
При наблюдении за больными с синдромом Марфана необходимо выполнять следующие требования к режиму труда, отдыха и реабилитации:
• детям с синдромом Марфана разрешаются занятия физкультурой только по ослабленной программе (спецгруппы и групы ЛФК);
• категорически запрещаются занятия в спортивных секциях, участие в соревнованиях, сельскохозяйственных работах, походах на длительные дистанции по пересеченной и горной местности, ношение тяжестей (не более 3 кг);
• категорически запрещены специальности, связанные с профессиональной вредностью: контакты с химическими веществами, лаками, красками, работа в условиях высоких температур и воздействия радиации, а также профессии, сопряженные с вибрацией, требующие высокой остроты зрения, больших физических и эмоциональных затрат;
• при выборе места жительства больным противопоказаны жаркий климат и зоны повышенной радиации;
• беременным женщинам с синдромом Марфана необходимо один раз в 2 мес проводить эхокардиографию. При диаметре аорты 45 мм и выше следует безотлагательно решать вопрос о целесообразности дальнейшего сохранения беременности;
• родоразрешение женщин с синдромом Марфана необходимо осуществлять с помощью кесарева сечения в специализированных родильных домах для рожениц с патологией сердечно-сосудистой системы.
Профориентация: больным с синдромом Марфана следует рекомендовать выбор профессий, не связанных с физическим перенапряжением (гуманитарные - юрист, педагог, экономист, биолог и пр.).
Профилактика. Больным с синдромом Марфана, вступающим в брак, показано медико-генетическое консультирование - информация о степени повторного риска развития у детей аналогичного заболевания. Наряду с этим, необходима также пренатальная диагностика.
4.2.2. Синдром Элерса-Данлоса
Синдром Элерса-Данлоса объединяет генетически гетерогенную группу поражений соединительной ткани. По утверждению V.A.McKusick, первый очевидный случай синдрома Элерса-Данлоса описан в 1682 году хирургом из Амстердама J.Van Meckeren. Автор наблюдал 17-летнего юношу, имевшего необычную растяжимость кожи. Почти два века спустя после первых сведений о синдроме Элерса-Данлоса, в 1892 г., русским дерматологом А.Н.Черногубовым было опубликовано сообщение о двух больных с этим заболеванием. Автор обратил внимание на тяжелое поражение кожи и суставов у пациентов и расценил данное состояние как генетически детерминированную «задержку вообще всей соединительной ткани».
|
|
|
