 |


Дифференциация популяций по грузу наследственных болезней 23 глава
|
|
|
|
Особое внимание уделяется назначе нию лекарственных средств, способствующих улучшению показателей клеточной биоэнергетики, перекисного окисления липидов и антиокислительной активности плазмы витаминов группы В, С и Е, рибоксина, коэнзима Qio, препаратов янтарной кислоты, цито-Мака, L-форм карнитина, пирацетама
Больным с синдромом Элерса-Данлоса показаны физиотерапевтические процедуры магнитотерапия на суставы, электро- сон, лечебная физкультура с преимущественным воздействием на позвоночник и суставы Рекомендуется также ежегодное пребывание (курс лечения 24 дня) в санаториях для больных с нарушениями функций опорно-двигательного аппарата или сердечно-сосудистой системы
Оперативное лечение включает торакопластику (по строгим показаниям), грыжесечение, операции на суставах, позвоночнике (также по строгим показаниям) и органе зрения Необходимо также не менее двух раз в год санировать хронические очаги инфекции полости рта и зубов
Опыт показывает, что примерно у 80-85% детей с синдромом Элерса-Данлоса проводимая терапия способствует улучшению или стабилизации основного патологического процесса [28]
Критериями эффективности назначенной терапии следует считать улучшение общего состояния ребенка (повышение толерантности к физической нагрузке, уменьшение частоты и интенсивности головных болей, увеличение мышечной силы и тонуса), сокращение числа спонтанных вывихов суставов, прекращение про- грессирования сколиоза, уменьшение геморрагического синдрома, стабилизацию степени пролабирования клапанов сердца, увеличение сократительной способности миокарда и улучшение показателей сердечного ритма (по данным ЭКГ, эхокардиографии и холтеровского монитори- рования) [28]
|
|
|
Улучшение общего состояния ребенка сопровождается положительной динамикой результатов лабораторных методов исследования крови снижением уровней лактата и пирувата, увеличением макроэргов (АТФ, АДФ, АМФ), нормализацией процессов перекисного окисления липидов (снижение малонового диальдегида и гидроперекисей), увеличением показателей антиокислительной активности плаз мы [43]
При наблюдении за больными с синдромом Элерса-Данлоса целесообразно выполнять следующие требования к режиму труда, отдыха и реабилитации
• детям с синдромом Элерса-Данлоса рекомендуются занятия физкультурой лишь по ослабленной программе (подготовительные и специальные группы и программы ЛФК) с исключением соревнований и занятий в спортивных секциях Следует также оберегать больных от сельскохозяйственных работ, походов на длительные дистанции, особенно по пересеченной и горной местности, ношения тяжестей (не более 3 кг),
• рекомендуется избегать профессиональных вредностей контактов с химическими и радиоактивными веществами, лаками, красками, работ в условиях высоких температур, а также сопряженых с большими физическими и эмоциональными затратами,
• нежелательно проживание больных в условиях жаркого климата и зонах повышенной радиации,
• родоразрешение женщин с синдромом Элерса-Данлоса целесообразно проводить в специализированных родильных домах для больных с патологией сердечнососудистой системы;
• больным с синдромом Элерса-Данлоса, вступающим в брак, необходима информация о степени повторного риска в семье аналогичного заболевания (медико- генетическое консультирование).
4.2.3. Туберозный склероз
Туберозный склероз - моногенное наследственное заболевание, характеризующееся полисистемным поражением нервной системы, кожи, внутренних органов, органов зрения, костной и нейроэндокрин- ной систем [44].
|
|
|
Заболевание впервые описано F. von Recklinghausen в 1862 г. как распространенный склероз [45]. В 1880 г. D-M.Bourneville у одного из детей с глубокой степенью умственной отсталости обнаружил своеобразные изменения в мозге, которые придавали извилинам узловатый вид, напоминавший клубни (тубер- сы). Для обозначения данного заболевания им предложен новый термин «туберозный склероз извилин мозга». Позже ретроспективно была детально проанализирована клиническая картина болезни В период новорожденности у ребенка возникли судорожные пароксизмы, выражавшиеся в заведении глазных яблок. В подростковом возрасте на лице больной было выявлено не наблюдаемое ранее клиницистами изменение кожи в виде «сливной везикулопапулезной сыпи на носу, щеках и лбу», которое D-M. Bourneville назвал «аспе rosacee». J.J.Prmgle в 1890 г. описал случай сходных кожных изменений у 25-летней женщины с легким снижением интеллекта. Кожные изменения он обозначил как «congenital adenoma cebaceum» (врожденная аденома сальных желез).
Туберозный склероз встречается с частотой 1:30 ООО населения Распространенность среди новорожденных варьирует от 1.5800 до 1: 10 000(46]
Генетические данные. Туберозный склероз наследуется по аутосомно-доми- нантному типу. Большинство случаев заболевания (80%) являются следствием мутации de novo. Болезнь отличается варьирующей экспрессивностью, почти 100% пенетрантностью и выраженной генетической гетерогенностью. Развитие туберозного склероза определяется двумя генами, локализованными в участке 34 длинного плеча хромосомы 9 (туберозный склероз I типа - TSC1) и в участке 13 короткого плеча хромосомы 16 (туберозный склероз II типа - TSC2). Мозаицизм туберозного склероза встречается только в тех случаях, когда часть клеток организма пациента содержит мутации в генах TSC1 или TSC2. Пациенты с мозаичным генотипом могут иметь полный спектр симптомов туберозного склероза [47, 48]. В этой связи в настоящее время выделяют два типа заболевания: туберозный склероз I типа и туберозный склероз II типа [49]
Клинические проявления. Основной симптомокомплекс туберозного склероза включает изменения кожи, внутренних органов, органов зрения, нервной и эндокринной систем.
Изменения кожи при туберозном склерозе довольно разнообразны и представлены гипопигментными пятнами, которые встречаются в 90% случаев и нередко обнаруживаются с рождения (рис. 4 2.3.1. на цветной вкладке). Число гипопигментных пятен варьирует от 3-4 до 100 и более [50]. Характерной особенностью является асимметричность их расположения. С младенчества могут выявляться белые пряди волос на голове, ресницах и бровях. В 15,4% случаев встречаются пигментные пятна цвета «кофе с молоком». Облигатными признаками туберозного склероза являются ангиофибромы лица (наблюдаются в 47-90% случаев у детей старше 4 лет) (рис. 4.2.3.2. на цветной вкладке), участки «шагреневой кожи» (в 21-68% случаев), околоногтевые фибромы и фиброзные бляшки (встречаются у 25% больных) (рис. 4.2.3.3 на цветной вкладке).
|
|
|
Мягкие фибромы встречаются у 30% больных. Они представляют собой множественные или единичные мягкие образования на ножках, мешотчатой формы, растущие на шее, туловище и конечностях (mol- luscum fibrosum pendulum). Другой вариант мягких фибром представляет собой множественные, несколько приподнятые над поверхностью кожи (и такого же цвета) мелкие образования, размером меньше булавочной головки, располагающиеся на туловище и шее и напоминающие гусиную кожу.
В стадии выраженных клинических проявлений в 45-66% случаев обнаруживаются костно-суставные изменения. При стандартном рентгенологическом исследовании трубчатых костей выявляются участки склероза, участки деструкции. В костях верхних и нижних конечностей обнаруживаются кисты, в плюсневых костях - периостальные дополнительные косточки, крайне редко - остеолиз костей. У большинства больных определяется остеопороз поясничного отдела позвоночника и трубчатых костей, что может быть обусловлено как длительным приемом антиконвульсантов, так и гормональными нарушениями [51, 52].
Примерно в половине случаев у больных туберозным склерозом встречаются поражения органов зрения в виде гамартом сетчатки и зрительного нерва [53]. Клинические проявления гамартом наблюдаются крайне редко. Основным симптомом является прогрессирующее снижение зрения.
|
|
|
Изменения внутренних органов включают разнообразные нарушения функции сердечно-сосудистой, пищеварительной, мочеполовой и других систем.
Изменения сердечно-сосудистой системы проявляются развитием рабдомиом (рис. 4.2.3.4.), которые, наряду с гипопиг- ментными пятнами, нередко являются первым клиническим признаком туберозного склероза [54,55]. Рабдомиомы встречаются в 30-60% случаев и чаще выявляются у лиц мужского пола. Они различаются по своим

Рис 4234 Множественные рабдомиемы сердца (ультразеукееее исследование).
формам и размерам, которые варьируют от нескольких миллиметров до нескольких сантиметров. Чаще всего опухоли не вызывают нарушений деятельности сердца. Клинически рабдомиомы сердца проявляются признаками сердечной недостаточности, нарушениями гемодинамики, сократительной функции миокарда и сердечного ритма. Они могут привести к смерти в пренаталь- ный и ранний натальный периоды.
Для рабдомиом сердца не характерно озлокачествление. В этой связи, оперативное вмешательство необходимо лишь в тех случаях, когда их размеры становятся значительными и они оказывают компрессионное воздействие на магистральные сосуды.
Сосуды средних и крупных размеров при туберозном склерозе поражаются крайне редко.
Изменения органов дыхания встречаются у 1% больных и наблюдаются чаще у лиц в возрасте старше 30 лет. Наиболее типичными поражениями легких являются кисты. Первыми клиническими симптомами туберозного склероза бывают дыхательная недостаточность и рецидивирующий пневмоторакс [56]. На рентгенограмме грудной клетки выявляется усиленный рисунок легочной паренхимы по типу «сотового легкого», распространяющийся на всю их паренхиму или только на ее изолированные участки.

Рис. 4 2.3.1 (к стр. 223). Гипопигмоитиыо пятна. Рис. 4.3.1.1 (к стр. 247). Синдром ломкой хромосомы X
(FRAXA) у ребенка.
Рис. 4.2.3.2 (к стр. 223). Ангиофибромы лица.
% I
Рис. 4.2 3.3 (к стр. 224). Околоиогтевыо фибромы.

Рис. 5.3 (к стр. 356,360). Молекулярно-цнтогенетнческая (FISH) диагностика (а) мозаичного случая синдрома Патау (видны ннтерфазные ядра с тремя н двумя хромосомами 13); (6) синдрома Дауна с помощью центромерных ДНК проб (слева - вндны три сигнала на хромосоме 21 н два на хромосоме 13) и сайт-сноцифичных ДНК-нроб на суб- теломерный участок хромосомы 21 (снрава - вндны три меченые хромосомы 21).
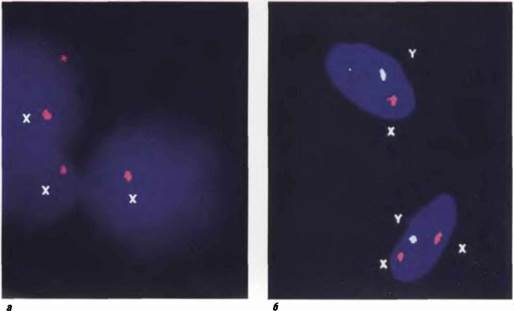
Рис. 5 7 (к стр. 363) (а) Молекулярно-цитогенетнческая (FISH) диагностика мозаичного случая синдрома Ulepeuiee- ского-Тернера 45,1/46,XX (видны два интерфазных ядра с одной хромосомой X н одно ннтерфазное ядро с двумя хромосомами X); (б) двухцвотовая ннтерфазная FISH при мозаичном случае синдрома Клайнфелтера 46,XY/47,XXY (хромосомы X маркированы красными сигналами и хромосома Y - светло-голубым сигналом).
|
|
|
Изменения в органах желудочно-кишечного тракта при туберозном склерозе разнообразны, встречаются относительно часто и проявляются патологией ротовой полости (узловые опухоли, фибромы или папилломы), печени (одиночные и множественные гамартомы и ангиомиолипомы, наблюдаемые у 10% больных) и прямой кишки (ректальные полипы, которые встречаются в 50-78% случаев [57, 58]. Ректальные полипы отличаются благоприятным прогнозом и, как правило, выявляются у больных старше 20 лет [59]. Клинически они бессимптомны, и лишь в отдельных случаях возможны боли при дефекации. Могут встречаться одиночные или множественные аденомы надпочечников, аденомы щитовидной железы [60], которые сопровождаются дисфункцией желез внутренней секреции.
В почках возникают множественные ангиомиолипомы и кисты (рис. 4.2 3.5.). Значительно реже встречаются карциномы. Почки поражаются у 40-80% больных [61, 62]. Поражение почек, как правило, выявляется во 2-3-м десятилетии жизни. Почечная недостаточность и карциномы наблюдаются у 5% больных с туберозным склерозом.
К характерным особенностям ренальных ангиомиолипом относятся их множественность и двусторонний (у '/3 больных) характер поражения. Клинически ангиомиолипомы длительное время не проявляются. Симптомы возникают, как правило, при увеличении массы и объема ангиомиолипом и проявляются развитием острых абдоминальных болей и шокового состояния, сопровождающегося падением артериального давления и вегетативными расстройствами.
Кисты почек при туберозном склерозе часто бывают небольшого размера, но могут достигать и нескольких сантиметров в диаметре. Обычно они рассредоточены в паренхиме и часто бессимптомны. Поликистоз почек особенно характерен для детей, тогда как ангиомиолипомы более типичны для взрослых. Кисты почек длительное время могут быть бессимптомными. Манифестными признаками выраженного поликистозно-

Рис 4 235 Ангиомиолипомы и кисты ночек (ультразвуковое иссиедоіание).
го повреждения почек являются артериальная гипертензия и умеренная азотемия [63].
Ангиомиолипомы и кисты почек обнаруживаются при ультразвуковом исследовании почек и компьютерной томографии.
Поражения нервной системы являются доминирующими в клинической картине туберозного склероза. Наиболее характерны судорожные пароксизмы, умственная отсталость, нарушения поведения, изменения в цикле «сон-бодрствование».
Судорожные пароксизмы развиваются у 80-92% больных [64-66] и часто начинаются на первом году жизни, в большинстве случаев - в первые месяцы.
Эпилептические пароксизмы нередко резистентны к противосудорожной терапии. P.Curatolo отмечено, что среди факторов, определяющих резистентность к противосудорожной терапии, наибольшее значение имеют дебют судорожных пароксизмов в возрасте до 1 года, наличие нескольких типов приступов, их высокая частота, трансформация тонических пароксизмов в парциальные [64].
Умственная отсталость при туберозном склерозе наблюдается в 48% случаев [67]. Степень умственной отсталости варьирует от умеренной до глубокой. Одной из главных причин, определяющих возникновение умственной отсталости, считаются судороги, возникающие на первом году жизни. Другой причиной могут быть корковые ту- берсы, локализующиеся в теменной, височной и лобной долях головного мозга. Нарушение интеллекта при туберозном склерозе сочетается с изменениями поведения в виде аутизма, гиперактивности, агрессивности
К ранним признакам аутизма у детей первого года жизни относится «безразличное отношение к состоянию комфорта» Обычно ребенок равнодушен к родителям, вяло реагирует на голос матери, кормление грудью Он практически не реагирует на обращенную к нему речь. В ряде случаев, родители, разочарованные эмоциональной вялостью ребенка, сами прекращают активную родительскую опеку и перестают с ним общаться.
В старшем возрасте ведущими признаками аутизма становятся нарушения коммуникации и «ригидность» поведения Коммуникационные проблемы заключаются, в первую очередь, в трудностях речевого общения Частым проявлением речевых нарушений, сопутствующих аутизму, бывает эхолалия (повторение слов и даже целых предложений непосредственно или через некоторое время после их произнесения). Диалог с детьми затруднен и нередко он протекает по типу «вопрос-ответ»
«Ригидность поведения» проявляется в виде патологической фиксации на каком- либо одном виде деятельности (навязчивое стремление следовать какому-либо одному маршруту, беседовать на одну тему). В то же время реальные ситуации, угрожающие жизни, часто недооцениваются пациентами.
Аутизм и гиперкинетическое поведение у детей с туберозным склерозом и выраженной степенью умственной отсталости связываются с наличием туберсов в лобных долях и задних отделах мозга [70].
В 60% случаев наблюдаются нарушения сна [71]
Наиболее типичными поражениями головного мозга при туберозном склерозе являются корковые туберсы, субэпенди- марные узлы и аномалии белого вещества мозга [72]
Корковые туберсы различаются по своим размерам, локализации, консистенции и форме Размер корковых туберсов варьирует от нескольких миллиметров до нескольких сантиметров Корковые туберсы располагаются в виде выступов над единичной или прилегающими бороздами Они расширяют борозду и сглаживают грань между серым и белым веществом Туберсы могут быть как единичными, так и множественными, имеют диффузную локализацию Каль- цификация туберсов отмечается в 54% случаев Число кальцифицированных туберсов увеличивается с возрастом больных
Наибольшую значимость в верификации туберсов при обследовании больных имеет магнитно-резонансная томография (МРТ), которая позволяет визуализировать туберсы в 95% случаев.
Субэпендимарные узлы встречаются в 95% случаев и выявляются как при компьютерной томографии, так и при МРТ мозга. Они локализуются, как правило, в стенках боковых желудочков, реже, в стенках III и IV желудочков мозга На компьютерных томограммах доминирующими признаками заболевания являются множественные полностью или частично кальци- фицированные субэпендимарные узлы округлой формы, локализующиеся в стенках боковых желудочков (рис 4 2 3 6, 4 2 3 7) Субэпендимарные узлы нередко трансформируются в гигантоклеточную астроцитому и выявляются у 10-15% больных [73, 74] Субэпендимарные ги- гантоклеточные астроцитомы манифестируют обычно между 5 и 10 годами жизни (средний возраст в момент выявления опухоли около 13 лет), обычно имеют тенденцию к росту и всегда локализуются у межжелудочкового отверстия Для диагностики гигантоклеточных астроцитом применяются как компьютерная томография, так и МРТ. В клинической практике нередко используют оба нейрофизиологических метода
Учитывая тот факт, что при туберозном склерозе гигантоклеточные астроцитомы
наблюдаются относительно часто, больным детям рекомендуется динамическое проведение нейрорадиологических исследований не реже одного раза в 2 года В случаях появления головных болей, рвоты, жалоб на ухудшение зрения необходимо экстренное проведение нейрорадиологических исследований.
Поражение белого вещества головного мозга характеризуется появлением своеобразных островков, состоящих из групп гетеротопических кластерных клеток и располагающихся вдоль линий, соединяющих эпендиму стенок желудочков и тубер- сы. Данные линии соответствуют нормальным миграционным путям спонгиобластов во время эмбриогенеза.
У 10% больных описаны поражения мозжечка [72].
В 1998 г. приняты диагностические критерии заболевания [49].
Первичные признаки:
• ангиофибромы лица или фиброзные бляшки на лбу;
• нетравматические околоногтевые фибромы;
• гипопигментные пятна (больше трех),
• участок «шагреневой кожи»;
• множественные гамартомы сетчатки;
• корковый туберс;
• субэпендимарные узлы;
• гигантоклеточная астроцитома;
• множественные или одиночные рабдомиомы сердца,
• лимфангиомиоматоз легких;
• множественные ангиомиолипомы почек
Вторичные признаки.
• многочисленные углубления в эмали
зубов;
• гамартоматозные ректальные полипы";
• костные кисты";
• миграционные тракты в белом веществе головного мозга;
• фибромы десен;
• гамартомы внутренних органов;
• ахроматический участок сетчатой оболочки глаза;
• на коже гипопигментные пятна по типу «конфетти»;
• множественные кисты почек".
а - требуется гистологическое подтверждение;
6 - достаточно рентгенологического подтверждения.

Рис 4 2 3 6 Магнитно-розонанснов исследование головного мозга (объяснения в тексте)

Рис 4237 Компьюторно-тамографичоскоо исследование головного мозга (объяснения в тексте)
Несомненный диагноз туберозного склероза устанавливают при наличии 2 первичных признаков или 1 первичного признака + 2 вторичных признаков
Возможный диагноз - 1 первичный признак + 1 или 2 (и больше) вторичных признака
Предположительный диагноз - или 1 первичный признак, или 2 (и больше) вторичных признака
Лечение. При туберозном склерозе лечение имеет преимущественно симптоматический характер
При эпилепсии подбор антиконвульсан- тов проводится с учетом характера судорожных приступов При лечении инфантильных спазмов препаратами выбора являются сабрил в дозе 40-150 мг/кг/сут (высокоэффективен в первые три года жизни и способствует ремиссии у 50-100% больных), препараты вальпроевой кислоты (де- пакин) из расчета 40-100 мг/кг/сут [75] При отсутствии эффекта рекомендуется сочетание вальпроевой кислотой с препаратами бензодиазепиновой группы или карбамазепинами
Для лечения парциальной эпилепсии препаратами выбора являются карбамазе- пины (тегретол, финлепсин и их ретарди- рованные формы) в дозе 15-40 мг/кг/сут В случае отсутствия эффекта монотерапии возможно сочетание их с ламикталом в дозе 0,5-10 мг/кг/сут Для лечения парциальных, а также генерализованных форм эпилепсий у детей старше 2 лет возможно применение комбинации препаратов вальпроевой кислоты с ламикталом в дозе 0,2-5 мг/кг/сут [76, 77]
Одной из самых сложных проблем при лечении больных с туберозным склерозом является коррекция умственной отсталости В связи с наличием судорожных пароксизмов применение ноотропных препаратов и стимулирующей терапии во многих случаях противопоказано Основной акцент при работе с умственно отсталыми пациентами делается на проведении ней- ропсихологической реабилитации [67]
Появление опухолей при туберозном склерозе ставит перед врачом проблему целесообразности оперативного лечения Как правило, тактика ведения больных выжидательная Хирургическое вмешательство показано лишь в случае быстрого роста опухоли, вызывающего нарушение функции органа
4.2.4. Несовершенное костеобразование
Несовершенное костеобразование
(osteogenesis imperfecta) - наследственное заболевание соединительной ткани, харак теризующееся множественными перело мами, остеопенией Заболевание впервые было выделено Лобштейном в 1825 г как самостоятельная форма под названием «Osteopsothyrosis idiopatica» В 1845 г Вро- ликом описана внутриутробная врожденная ломкость костей, названная им osteogenesis imperfecta
Частота заболевания среди новорожденных составляет 21,8 на 100 000, а во всей детской популяции -10,6 на 100 000 [78,79]
Гэнетические данные и патогенез. Несовершенное костеобразование I типа является доминантно наследуемым, генерализованным поражением соединительной ткани, характеризующимся, главным образом, ломкостью костей и голубыми склерами Заболевание связано с дефектом коллагена I типа - основного структурного белка соединительной ткани - костного матри- кса, кожи и сухожилий В большинстве случаев функциональный «нуль-аллель» гена COLIA1 на хромосоме 17 или гена COLIA2 на хромосоме 7 приводят к уменьшению образования нормального коллагена I типа Молекулярные дефекты при этом заболевании представлены различными типами мутаций (миссенс-мутации, сплайсинговые мутации, образование стоп-кодонов и др), которые приводят к неспособности про- альфа-1(1) цепей полноценно включаться в молекулу коллагена и определяют формирование различных фенотипов К сходному фенотипу приводят и мутации в гене цепи А2 коллагена типа I [80]
Возникновение заболевания, обусловленное мутациями, затрагивающими только образование отдельных цепей коллагена (А1 или А2), нередко сопровождается более легким течением, которое, по- видимому, связано с тем, что при этом продуцируется структурно нормальный коллаген, но в уменьшенном количестве [81, 82] При тяжелых формах имеются структурные дефекты цепей коллагена, приводящие к дезорганизации и ослаблению костного матрикса вследствие поражения соединительно-тканной основы кости [6] Несмотря на то, что коллаген I типа присутствует не только в костном мат- риксе, но также в коже и сосудистой стенке, несовершенное костеобразование - заболевание преимущественно костной системы, возможно, вследствие того, что мутантный тип коллагена в наибольшей степени образуется остеобластами и ак тивно включается в костный матрикс [82] С помощью биохимических исследований установлено, что при несовершенном ос- теогенезе изменяются свойства коллагена I типа, нарушается нормальное образование коллагена I типа (цепи А1 и А2) [79]
Показано, что у больных с несовершенным остеогенезом I типа в культуре фибробластов снижено образование прокол- лагена I
Пенетрантность голубых склер составляет почти 100%, в то время как снижение слуха носит возрастно-зависимый характер [84] Показано, что возникновение новых мутаций при этом заболевании может зависеть от возраста отца [85]
Заболевание может быть унаследованным, но довольно часто встречаются спорадические случаи, которые, вероятно, связаны с новыми мутациями («свежие» мутации) В ранее проведенных исследованиях частота доминантных мутаций гена несовершенного костеобразования оценивалась как 0,7 х 105 [86]
Классификация. Используя клинические, радиологические и генетические критерии, D О Sillence et al [82] выделили четыре типа заболевания I тип - ауто- сомно-доминантный с голубыми склерами - наиболее легкая форма патологии, проявляющаяся остеопенией и переломами при незначительной травме, умеренным отставанием в росте, II тип - перинатальный синдром с летальным исходом - проявляется относительно большим черепом с тонкими, мягкими костями свода черепа, короткими деформированными конечностями, узкой грудной клеткой, при рентгенографии трубчатых костей выявляются следы множественных внутриутробных переломов, III тип - форма заболевания с прогрессирующими деформациями скелета, особенно нижних конечностей, с нормальным цветом склеры глаз, IV тип - аутосомно-доминантный с нормальными склерами - характеризуется повторными переломами с деформациями конечностей и грудной клетки различной степени тяжести, пониженной двигательной активностью, кифосколио- зом, платиспондилией, задержкой роста Эта классификация является неполной (отдельные характеристики частично совпадают между собой) и в настоящее время дополняется новыми тремя типами [83] Разделение заболевания на врожденные и поздние формы в настоящее время оставлено При различных типах заболевания может также наблюдаться нарушение дентиногенеза
Клиническая картина. Несовершенное костеобразование отличается выраженным клиническим полиморфизмом Заболевание характеризуется множественными переломами, обычно в результате незначительных травм По числу переломов и степени инвалидности существует выраженная внутрисемейная и межсемейная вариабельность заболевания [87]
Сроки выявления заболевания также широко варьируют - от 20 нед внутриутробного развития до 2-3-летнего возраста Количество переломов также варьирует - от 1 до 50 и выше, при этом число переломов также колеблется - от одного в 1-2 года до 15-20 в течение года Пораженные дети имеют голубые склеры, нормальные зубы и нормальное или небольшое отставание в росте Однако могут иметь место и нарушения дентиногенеза Переломы редки в неонатальном периоде, но становятся постоянными от детского до пубертатного периода, когда частота их сначала снижа ется и вновь нарастает в период менопаузы у женщин и после 60 лет у мужчин Пе реломы быстро заживают с образованием достаточно хорошей костной мозоли без деформаций Снижение слуха наблюдается почти у 50% больных, оно обычно начинается к концу 10-летнего возраста и постепенно достигает степени тугоухости Помимо костной патологии у больных с несовершенным костеобразованием обнаруживается тонкая кожа, с низкой эластич ностю Кожа обычно натянута, ригидна, имеет синюшный оттенок [88] Склеры у больных с I типом несовершенного косте образования обычно голубые в течение всей жизни, в отличие от II и III типов, при которых склеры могут быть голубыми или при рождении, или у грудных детей Интенсивность окраски склер может варьировать от серо голубой до почти полного обесцвечивания к подростковому возрас ту Встречаются и другие изменения со стороны органа зрения (шаровидный хрусталик, маленький диаметр роговицы малая подвижность глаз, старческая дуга и др) Нередко выявляются макроцефалия, треугольное лицо, умеренная гиперпо движность в суставах, кифосколиоз, гры жи В разные возрастные периоды могут выявляться изменения со стороны органов кровообращения пролапс митрального клапана (у 18% больных часто без регур- гитации), недостаточность аортальиых клапанов и легкое расширение корня аорты (примерно у 12% больных), стеноз аор ты, который не прогрессирует при применении бета адреноблокаторов У 59 5% больных с несовершенным костеообразо- ванием наблюдается снижение слуха, ко торое выявляется чаще всего у детей со второй декады жизни [89] Снижение слуха может быть как по кондуктивному, так и по нейросенсорному типу С отосклерозом часто ассоциируется головокружение
При рентгенологическом исследовании костей черепа находят вормиевы косточки, но при рождении ребенка морфология кос ти обычно нормальная, хотя могут иметь место умеренная остеопения и искривление бедренных костей Морфология позво ночника часто также нормальная, но не редко формируются «рыбьи» позвонки (выявляемые при рентгенологическом исследовании) Возможна молекулярная пренатальная диагностика заболевания [90]
Лечение больных с несовершенным костеобразованием проводится с целью минимизировать переломы и максимизировать функции, то есть предполагает уменьшение частоты переломов и увели чение двигательной активности больных Используются ортопедическое - хирургическое и консервативное - лечение пере ломов а также лекарственная терапия Последняя до сих пор разработана недос таточно Медикаментозные средства включают препараты кальция, кальцито- нин, дифосфонаты Изолированное при менение фторидов и монотерапия кальци- тонином признаны малоэффективными [91-93] При тяжелых формах заболева ния (III и IV типы, по D Sillence) с успехом применяется памидронат (аминогидрок- сипропилиден бифосфонат), который яв ляется синтетическим аналогом пирофос фата, оказывающего ингибирующее действие на остеокластическую резорбцию кости [94, 95] Препарат вводится внутри венно в дозе 1 5-6 мг/кг массы тела в год с 4-6-месячными интервалами Лечение памидронатом в течение 22-29 мес приводило к уменьшению числа переломов до 1-2 в год и увеличению плотности ко стной ткани на 20-60% за счет тормозя щего влияния на остеокластические про цессы [91, 96] Отмечены уменьшение ак тивности щелочной фосфатазы содержания N-телопептида в сыворотке крови.
|
|
|
