 |


Дифференциация популяций по грузу наследственных болезней 25 глава
|
|
|
|
16 Блинникова ОЕ, Курникова МА, Мутовин ГР Клиника классификация, диагностика синдрома Эперса-Данлоса в свете современных молекулярно-генетических исследований Новый хирургический архив 2002 1(4) (Интернет-журнал- Surgeon spb ru)
17 McKusick VA Mendelian Inheritance in Man NY, 1993
18 Steinmann В, Royce P M, Superti-Furga A The Ehlers-Danlos syndrome In P M Royce, В Steinmann, eds Connective Tissue and its Heritable Disorders Molecular Genetic and MediLal Aspects N Y Wiley-Liss Ins, 1993,351-407
19 Prockop D J, Kivinkko КI Collagens molecular biology, diseases, and potentials therapy Ann Rev Biochem 1995 64 34-43
20 Hamel В С J, Pals G, Engels С H, et al Ehlers-Danlos syndrome and type III collagen abnormalities a variable clinical spectrum Clin Genet 1999 82 305-11
21 Loughlin J, Irven С, Hardwick L J, et al Linkade of the gene that encodes the alpha -1 chain of type V collagen (COL5A1) to type II Ehlers-Danlos syndrome (EDS II) Hum Molec Genet 1995, 4 1649-51
22 Michalickova К Susis M, Willing M С, et al Mutations of the a2 (V) chain of type V collagen impair matrix assembly and produce Ehlers-Danlos syndrome type I Hum Molec Genet 1998, 7(5) 249-55
23 Barabas A P Heterogeneity of the Ehlers-Danlos syndrome description of three clinical types and hypothesis to explain the basis defect(s) Br Med J 1967 2(552) 612-3
24 Beighton P The Ehlers-Danlos syndrome London William Heinemann (pub) 1970
25 Beighton P, De Paepe A Steinmann В, et al Ehlers-Danlos Syndromes Revised Nosology, Villefranche, 1997 Am J Med Genet 1998, 77 31-7
26 SchapiraAH Mitochondrial disorders//Biochem Biophys Acta 1999 1410 99-102
27 Бураковский В И, Бокерия Л А Сердечнососудистая хирургия М, 1989, 608-27
28 Тернова Т И, Бочкова Д Н Изменение сердечно-сосудистой системы при синдроме Элер- са-Данлоса Вестник АМН СССР 1984, (2) 65-8
29 Nuyting L Freund М, Lagae L, et al Classical Ehlers-Danlos Syndrome Caused by a Mutation in Type I Collagen Am J Hum Genet 2000, 66 1398-402
30 SokoIov В P etal Exclusion of COLIA1, COLIA2 and COLIIIA1 genes as candidate genes for Ehlers-Danlos syndrome type I in one large family Hum Genet 1991, 88 125-9
31 Birk D E, Fitch J M, Babiars J P, et al Collagen fibrillogenesis in vitro interaction of types I and V collagen regulates fibril diameter J Cell Sci 1990,95 649-57
32 Burrows N P et al The gene encoding collagen alpha-1 (V) (COL5A1) is linked to mixed Ehlers-Danlos syndrome type l/ll J Invest Derm 1996,196 1273-6
33 Richards A J et al A single base mutation in COL5A2 causes Ehlers-Danlos syndrome type II J Med Genet 1998,35 846-8
34 Burch G H, Gong Y, Liu W, et al Tenascin-X deficiency is associated with Ehlers-Danlos syndrome Nature Genetics 1997,17 104-8
|
|
|
35 Narcisi P, Richards A J, Ferguson S D, Pope F M A family with Ehlers-Danlos syndrome type III/ articular hypermobility syndrome has a glycine 637-to-serine substitution in type III collagen Hum Molec Genet 1994, 3 1617-20
36 Viljoen D, Goldblatt J, Thompson D, Beighton P Ehlers-Danlos syndrome yet another type? Clin Genet 1987,32 196-291
37 Gilchrist D, Schwarze U, Shields К, et al Large kindred with Ehlers-Danlos syndrome type IV due to a point mutation (G571S) in the COL3A1 gene of type III procollagen low risk of pregnancy complications and unexpected longevity in some affected relatives Am J Med Genet 1999,82 305-11
38 Steinmann В, Superti-Furga A, Joller-Jemelka H I, et al Ehlers-Danlos syndrome type IV - a subset of patients distinguished by low serum levels of the amino-terminal propeptide of type III procollagen Am J Med Genet 1989, 34 68-71
39 Pope F M, Nicholls А С Pregnancy and Ehlers-Danlos syndrome type IV (Letter) Lancet 1983,1 249-50
40 Rudd N L, Nimrod С, Holbrook К A Byers P H Pregnancy complications in type IV Ehlers-Danlos syndrome Lancet 1983,1 50-3
41 Вельтищев Ю E Казанцева Л 3, Семячкина А Н Наследственная патология человека Под ред Ю Е Вельтищева, Н П Бочкова М, 1992,1 91-120
42 Howell N Human mitochondrial diseases answering questions and questioning answers Int Rev Cytol 1999,186 49-116
43 De Vivo D The expanding spectrum of mitochondrial diseases Brain Develop 1993, 15 1-22
44 Бадалян Л 0, Тоболин В А, Вельтищев Ю Е Наследственные болезни у детей М Медицина, 1971, 318-21
45 Gomez М R History of Tuberous Sclerosis Complex In Tuberous Sclerosis M Gomes, J Sampson, V Whittemore, ed N Y -Oxford Oxford University Press, 1999, 3-9
46 Osborne J P, Fryer A, Webb D Epidemiology of Tuberous Sclerosis Ann NY Acad Sci 1991, 615 125-8
47 Sampson J R The TSC2 Gene and Tuberin In Tuberous Sclerosis Ed M Gomes, J Sampson, V Whittemore NY-Oxford Oxford University Press, 1999,188
48 Kwiatkowski D J The TSC1 Gene Identification, mutations and Mosaicism In Tuberous Sclerosis M Gomes, J Sampson, V Whittemore, ed NY-Oxford Oxford University Press, 1999, 275-87
49 Roach E S, DiMario F J, Kandt R S, Northrup H Tuberous Sclerosis Consensus Conference Recommendations for Diagnostic Evaluation J Child Neurol 1999,14 401-7
50 Rogers R S, O'Connor W J Dermatologic Manifestations In Tuberous Sclerosis M Gomes, J Sampson, V Whittemore, ed N Y -Dxford Oxford University Press, 1999,160-80
51 Morozov A, Lapkina S, Dorofeeva M, et al Skeletal involvement in Tuberous sclerosis TSC International Research Symposium '96 11-13 September 1996 Bath, UK
52 Hoffman AD Imaging of Skeleton and Great Vessels In Tuberous Sclerosis Ed M Gomes, J Sampson, V Whittemore N Y -Oxford Dxford University Press, 1999, 240-9
53 Robertson D M Ophthalmic Findings In Tuberous Sclerosis M Gomes, J Sampson, V Whittemore, ed NY-Oxford Oxford University Press, 1999,145-59
54 Mair D D, Edwards W D Seward J В Cardiac Manifestations In Tuberous Sclerosis M Gomes, J Sampson, V Whittemore, ed N Y - Oxford Oxford University Press 1999 194-206
55 Белозеров Ю M, Дорофеева M Ю, Березниц- каяВВ идр Опухоли сердца при туберозном склерозе Современные инвазивные и неин- вазивные методы диагностики Ультразвук электрофизиология М АИРАРТ, 2000 132-6
|
|
|
56 Castro М, Shepherd С W, Gomez М R, et al Pulmonary tuberous sclerosis Chest 1995 107 189-95
57 Gomez M R Liver, Digestive Tract, Spleen Arteries, Thymus and Lymphatics In Tuberous Sclerosis M Gomes, J Sampson, V Whittemore, ed NY-Oxford Oxford University Press, 1999, 228-39
58 Strotzer M, Paetzel С, Feuerbach S Multiple hepatic angiolipomas a case report and review of literature Eur Radiol 1999, 9 259-61
59 Gould S R Gamartomas rectal polyps are common in tuberous sclerosis Anne NY Acad Sci 1991,615 71-80
60 Zimmerman D The endocrine system in tuberous sclerosis complex In Tuberous Sclerosis M Gomes, J Sampson, V Whittemore, ed N Y - Oxford Oxford University Press, 1999, 218-27
61 Катышева О В, Игнатова М С, Дорофеева М Ю и др Почечные синдромы при туберозном склерозе Практическая нефрология 1997, 3 20-6
62 Bjornsson J, Henske Е Р, Bernstein J Renal Manifestations In Tuberous Sclerosis M Gomes, J Sampson, V Whittemore, ed N Y -Oxford Oxford University Press, 1999,181-93
63 Stiilwell T J, Gomez M R, Kelalis P P Renal lesions in tuberous sclerosis J Urol 1987,138 477-81
64 Curatolo P Neurological aspects of Tuberous Sclerosis Complex TSC International Research Symposium 96 11-13 September 1996 Bath, UK

Рис. 7.1.2 (к стр. 390). Расширенные первые пальцы кистей у ребеика с синдромом Рубииштейиа-Тейби,

Рис. 7.11 (к стр. 390). Девочка, 1 г. 7 мес, с синдромом Рубинштейна-Тейби. Приподнятые дугообразные брови, антимонголоидный разрез глаз, зпикант, гримаса, напоминающая улыбку.
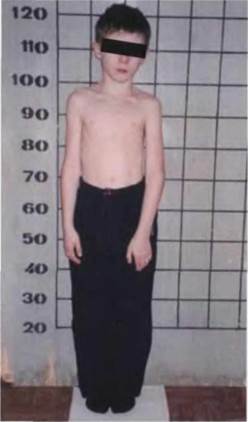
Рис. 7.1.4 (к стр. 397). Мальчик, 10 лет, с синдромом Сильвера-Рассела. Низкий рост, «треугольное» лицо, асимметрия скелета.
Рис. 71.5 (к стр. 400) Мальчик, 8 лет, с синдромом Ну- иаи. Антимонголоидный разрез глаз, зпикантус, птоз еек, низкорасположениые ушные раковины.

Рис. 7.1.3 (к стр. 394). Мальчик, 2 г. 6 мес, с синдромом Вильямса. «Лицо зльфа»: зпикантус, вывернутые вперед ноздри, маленькая нижняя челюсть, опущенные вниз щеки, большой рот, «звездчатый» рисуиок радужки.

Рис. 7.1.7 (к стр. 406) Мальчик, 1 г. 9 мес, с синдромом Секкеля. Микроцефалия, узкое лицо, маленькая нижняя челюсть, выступающий клювовидный нос, большие глаза, увеличенные ушные раковины.

Рис. 7.2.1 (к стр. 410). Мальчик, 13 лет, с синдромом Бе- кеита-Вндемаиа. Высокие показатели физического развития.
| ) | Г і | ||
| г | |||
| г | 1 1 I |
s
Рис. 7.1.6 (к стр. 400) Мальчик, 8 лет, с синдромом Ну- иан. Щитообразная форма грудной клетки, гилертело- ризм сосков.

Рис. 7.2.3 (к стр. 414) Девочка с синдромом Вивера: высокие показатели физического развития, макроцефалия, косоглазие, умственная отсталость.
|
|
|
65 Rnlkonen R Simell 0 Tuberous Sclerosis and Infantile Spasms Develop Med Child Neurol 1990 32 203-9
66 Curatolo P Tuberous Sclerosis In Infantile Spasms and West Syndrome Ed 0 Dulac, HChugam BDalla Bernandina London, Philadelphia, Toronto, Sydney, Токіо W В Saunders Company Ltd 1994,192-202
67 Hunt A Psychiatric and Psychological Aspects In Tuberous Sclerosis Ed M Gomes, J Sampson, VWhittemore NY-Oxford Oxford University Press, 1999,47-62
68 Bolton P Cognitive and Behavioural Developments in Tuberous Sclerosis TSC Millennium Research Symposium 2000 «From genes to treatment», 13-15 September 2000 Edinburgh, Scotland
69 Gillberg I С, Gillberg С, Ahlsen G Autistic behavior and attention deficits in tuberous sclerosis a population-based study Develop Med Child Neurol 1994, 36 50-6
70 Curatolo P. Cusmai R, Cortesi F, et al Neuropsychiatric aspects of tuberous sclerosis Ann NY Acad Sci 1991,615 8-16
71 Hunt A, Stores G Sleep disorder and epilepsy in children with tuberous sclerosis a questionnaire-based study Develop Med Child Neurol 1994 36 108-15
72 Inoue Y Nemoto Y, Murata R, et al CT and MR imaging of cerebral tuberous sclerosis Brain Develop 1998 20 209-21
73 Shepherd С W, Scheithauer В W Gomez M R, et al Subependymal Giant Cell Astrocytoma A Clinical, Pathological, and Flow Cytometric Study Neurosurg 1991, 28 864-8
74 Torres О A, Roach E S, Delgado MR et al Early Diagnosis of Subependymal Giant Cell Astrocytoma in Patients With Tuberous Sclerosis J Child Neurol 1998 13 173-7
75 Hancock E Osborne J P Vigabatrin in treatment of infantile spasms in tuberous sclerosis J Child Neurol 1999,14 71-4
76 linuma К General principles of treatment and effects of childhood intractable epilepsy Rinsho Shin Keigaku 1999 39 75-6
77 Schapel G J Wallace S J Gordon G S A survey of lamotrigme and vigabatrin treatment in children with severe epilepsy Seizure 1997, 6 479-83
78 Andersen P E, Hauge M Osteogenesis imperfecta a genetic, radiological, epidemiological study Clin Genet 1989, 36 250-5
79 Byers P H Osteogenesis imperfecta In P M Royce В Steinmann Connective tissue and its heritable disorders molecular, genetic, and medical aspects NY Wiley-Liss(pub), 1993, 317-50
80 Zhuang J, Tromp G, Kuivaniemi H, et al Deletion of 19 base pairs in intron 13 the gene for the pro-alpha-2(1) chain of type 1 procolla- gen(COL1A2) causes exon skipping in a proband with type 1 osteogenesis imperfecta Hum Genet 1993,91 210-6
81 Reing С M Report on new types of intramedullary rods and treatment effectiveness data for selection of intramedullary rodding in osteogenesis imperfecta Connect Tiss Res 1995, 31 (Suppl) 77-9
82 Sillence D 0, Senn A, Danks D M Genetnc heterogeneity in osteogenesis imperfecta J Med Genet 1979,16 101-16
83 Willing M С, Deschenes S P, Scott D A, et al Osteogenesis imperfecta type 1 molecular heterogeneity for COL1A1 null alleles of type 1 collagen Am J Hum Genet 1994, 55 638-47
84 Garretsen T J T M, Cremers С W R J Clinical and genetic aspects in autosomal dominant inherited osteogenesis imperfecta, type 1 Ann NY Acad Sci 1991,630 240-8
85 Carothers A D, McAllion S J, Paterson С R Risk of dominant mutation in older fathers evidence from osteogenesis imperfecta Med Genet 1986, 23 227-30
86 Smars G Osteogenesis imperfekta in Sweden Clinical Genetic, Epidemiological and Socio- medical Aspects Svenska Borforlagert Norstedts Stockholm, 1961
87 Rowe DW, Shapiro JR, Schlesinger S Diminished type 1 collagen synthesis and reduced alphal (1) collagen messenger RNA in cultured fibroblasts from patients with dominant- ly inherited (type 1) osteogenesis imperfecta J Clin Invest 1985 76 604-11
|
|
|
88 Hansen В Jemec G В E The mechanical properties of skin in osteogenesis imperfecta Arch Derm 2002,138 909-11
89 Kuurila К, Kentala E, Karjalainen S, et al Vestibular dysfunction in adult patients with osteogenesis imperfecta Am J Med Genet 2003,120A 35-358
90 De Vos A, Sermon К, Van de Velde H, et al Two pregnancies after preimlantation genetic diagnosis for osteogenesis imperfecta, type 1 and type 1V Hum Genet, 2000,106 605-13
91 Manni J С Gerber N L Osteogenesis imperfecta rehabilitation and prospects for gene therapy JAMA 1997, 277 746-50
92 Marini J С Osteogenesis imperfecta comprehensive management Adv Pediatr 1988, 35 391-426
93 Procop D J, Kivirkko КI Collagens molecular biology, diseases and potentials for therapy Ann Rev Biochem 1995, 64 403-34
94 Arikoski P, Silverwood B, Tillman V, et al Intravenous pamidronate treatment in children with moderate to severe Osteogenesis imperfecta assessment of indices of dualenergy X-ray absorptiometry and bone metabolic markers during the first year of therapy Bone 2004,34(3) 539-46
95 Marini J С Osteogenesis imperfecta - managing brittle bones (Editorial) New Eng J Med 1998 339 986-7
96 Bembi В, Parma A, Bottega M, et al Intravenous pamidronate treatmelnt in osteogenesis imperfecta J Pediat 1997,131 622-5
97 Lee Y -S Low S -L, Lim L -A, et al Cyclic pamidronate infusion improves bone mineralisation and reduces fracture incidence in osteogenesis imperfecta Eur J Pediatr 2001,160 641-4
98 Glorieux F H, Bishop N J, Plotkin H. et al Cyclic admistration of pamidronate in children with severe osteogenesis imperfecta New Eng J Med 1998, 339 947-52
99 Zeitlin L, Rauch F, Plotkin H, et al Height ana weight development during four years of therapy with cyclical intravenous pamidronate in children and adolescents with osteogenesis imperfecta types 1,111 and 1V Pediatr 2003,111 1030-6
100 Rauch F, Plotkin H, Travers R, et al Osteogenesis imperfecta types 1111 and 1V effect of pamidronate therapy on bone and mineral metabolism J Clin Endocr Metab 2003,88 986-92
101 Lindsay R Modeling the benefits of pamidronate in children with osteogenesis imperfecta J Clin Invest 2002 110 1239-41
102 Бережной А П Снетков А И Белова H А Комплексное лечение несовершенного остеогене- за Вопросы травматологии и ортопедии 1989 (12) 52-5
103 Вельтищев ЮЕ, Барашнев ЮИ, Казанцева Л 3, Белова Н А Об эффективности лечения детей с наследственной патологией в специализированной клинике Педиатрия 1990,54-61
104 Niyibizi С, Wang S, Mi Z, et al Gene therapy approaches for osteogenesis imperfecta 2004, 11(4) 408-16
105 Huson S M Compston DAS, Clark P, et al A genetic study of von Recklinghausen neurofibro matosis in south east Wales 1 Prevalence, fitness mutation rate, and effect of parental transmission on severity J Med Genet 1989 26 704-11
106 Barker D, Wright E, Nguyen К et al The gene for NF1(von Recklinghausen neurofibromatosis) is on chromosome 17 near the centromere Cytogenet Cell Genet 1987 46 576
107 ColmanSD RasmussenSA HoV etal Somatic mosaicism in patient with neurofibromatosis type 1 Am J Hum Genet 1996 58 484-90
108 Shen M N, Harper P S UpadhyayaM Molecular genetics of neurofibromatosis type 1 (NF1) J Med Genet 1996 33 2-17
109 Karvonen SL, Koivunen J, Nissmen M, et al Neurofibromatosis type 1 tumor suppressor gene expression is deficient in psoriac skin in vivo and in vitro potential link to increased ras activity Br J Dermatol 2004,150(2) 211-9
110 Gutman DH, Wu YL Hedrick NM et al Heterozygosity for neurofibromatosis 1 (NF1) tumor suppressor results in abnormalities in cell attachment, spreading and motility in astrocytes Hum Molec Genet 2001,10 3009-16
111 RaggeNK FalkRE Cohen WE, etal Images if Lisch nodules across the spectrum Eye 1993, 7 95-101
112 SkuseG R, Kosciolek В A, Rowley P T Moltcular genetic analysis of tumors in von Recklinghausen neurofibromatosis loss heterozygosity for chromosome 17 Genes Chromosomes Cancer 1989,1 36-41
113 LegiusE DescheemaekerMJ FrynsJP etal Neurofibromatosis type 1 Genet Counsel 1994 5 225-41
114 Arun 0 Gutmann О H Recent advances in neurofibromatosis type 1 Curr Opin Neurol 2004, 17(2) 101-5
115 Konishi K, Nakamura M, Yamakawa H, et al Case report hypophosphatemic osteomalacia in von Recklinghausen neurofibromatosis Am J Med Sci 1991, 301 322-8
|
|
|
116 Босин В Ю, Кондрина В В, Табакова Л И и др Рентгенологические изменения скелета при нейрофиброматозе у детей Мед радиол и радиационная безопасность 1997, (6) 15-9
117 Leqius М J, Descheemaeker М J, Spaepen А, et al Neuropsychological profile in children with NF1 Genet Counsel 1994,5 213-4
118 Seizinger В R, Martuza R L, Gusella J F Loss of genes on chromosome 22 in tumori genesis of human acoustic neuroma Nature 1986 322 644-7
119 Wertelecki W, Rouleau G A, Superneau D W, et al Neurofibromatosis 2 clinical and ONA linkage studies of a large kindred New Eng J Med 1988, 319 278-83
120 Watson С J, Gaunt L, Evans G, et al A disease associated germline deletion maps the type 2 neurofibromatosis(NF2) gene between the Ewing sarcoma region and leukaemia inhibitory factor locus Hum Molec Genet 1993, 2 701-4
121 Evans DGR, Trueman L, Wallace A, et al Genotype/phenotype correlations in type 2 neu- rofibromatosis(NF2) evidence for more severe disease associated with truncating mutations J Med Genet 1998, 35 450-5
122 Parry 0 M, MacCollin M M, Kaiser-Kupfer M I. et al Germ-line mutations in the neurofibromatosis 2 gene correlations with disease severity and retinal abnormalities Am J Hum Genet 1996,59 529-39
123 Evans 0 G, Huson S M, Oonnai 0, et al A clinical study of type 2 neurofibromatosis Quart J Med 1992, 84 603-18
124 Ragge N К, Baser M E. Klein J, et al Ocular abnormalities in neurofibromatosis 2 Am J Ophthal 1995,120. 534-641
125 Parry D M, Eldridge R, Kaiser-Kupfer MI, et al Neurofibromatosis 2 (NF2) clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity Am J Med Genet 1994, 52 450-61
126 Martuza RL, Eldridge R Neurofibromatosis 2 (bilateral acoustic neurofibromatosis) New Eng J Med 1988, 318 684-8
127 Gutman D H, Aylsworth A, Carley J, et al The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 11 JAMA 1997, 278 51-7
128 Темин П A, Табакова Л И Диагностика и дифференциальная диагностика нейрофиброматоза, тип I у детей Вестник практикующего невролога 1997, (3) 128-37
129 Hirokawa Y, Tikoo А, Huynh J, et al A clue to the therapy of neurofibromatosis type 2 NF2/ merlin isaPAKI inhibitor Cancer 2004,10(1) 20-6
130 Lee V, Ragge N К. Collin J R Orbitotemporal neurofibromatosis Clinical features and surgical management 2004,111(2) 382-8
4.3. Моногенные болезни, имеющие сцепленный с Х-хромосомой тип наследования
Общая характеристика Х-сцепленных (доминантных и рецессивных) болезней
В связи с тем, что с Y-хромосомой практически не наследуется ни одно серьезное заболевание, сцепленное с полом наследование почти в 100% случаев означает сцепление с Х-хромосомой Эти заболевания занимают более заметное место в практике медико-генетического консультирования, чем можно было бы ожидать, если исходить из относительного вклада Х-хромосомы в геном человека
К настоящему времени известно более 850 Х-сцепленных заболеваний и наследственных фенотипов Подавляющее их большинство относится к Х-сцепленным рецессивным, намного меньше - к Х-сцепленным доминантным и еще меньше - к доминантным с летальным исходом для гемизиготных плодов мужского пола. По отношению к
Х-сцепленным заболеваниям термины «доминантность» и «рецессивность» следует применять с осторожностью, так как у гетерозиготных женщин наблюдается ббльшая степень вариабельности по сравнению с ау- тосомными заболеваниями. 6 значительной мере это связано с результатами инактивации Х-хромосомы, которая затрагивает исключительно одну из Х-хромосом у женщин в раннем эмбриональном периоде Различная степень вариабельности в инактивации Х-хромосомы проявляется в виде более мягкой и вариабельной симптоматики наследственной патологии.
Распознавание Х-сцепленного характера наследования заболевания по данным родословной имеет принципиальное значение для определения медико-генетического прогноза, однако следует подчеркнуть, что этот тип наследования удивительно часто пропускается. Для педиатра крайне необходимо знание основных признаков Х-сцепленных заболеваний
Наиболее характерными чертами Х-сцв- пленных признаков и болезней являются следующие
1 Зависимость клинических проявлений от того, является ли признак рецессивным или доминантным
2 При Х-сцепленном рецессивном типе наследования (гемофилия, некоторые формы миодистрофий и др) гетерозиготы фенотипически здоровы Чаще всего в данном случае речь идет о женщинах-носительницах, так как в норме они несут две Х-хромосомы У женщин развивается заболевание в том случае, если мутацию несут обе Х-хромосомы или имеет место дефицит одной Х-хромосомы (кариотип 45,X) В основном заболевания с подобным типом наследования проявляются у мужчин, так как у таких мужчин патологический ген проявляет свое действие в ге- мизиготном состоянии (чаще всего больные рождаются в браке женщин-гетерози- гот и здоровых мужчин)
3 При Х-сцепленном доминантном типе наследования (фосфат-диабет и др) фено- типические проявления будут иметь как ге- мизиготы, так и гетерозиготы
Все сыновья и их дети-мальчики в браке здоровой женщины и больного мужчины будут здоровы, так как отец может им передать только Y-хромосому Все дочери будут облигатными носителями (гетерозиготы) и фенотипически больными
Передачи заболевания от мужчины к мужчине не наблюдается, так как сын никогда не наследует Х-хромосому от отца
Дочери больного мужчины, получившие патологический ген от отца, становятся больными, если наследование Х-сцеплен- ное доминантное, или будут носительницами заболевания, если наследование Х-сце- пленное рецессивное
4 Степень риска для сыновей женщины-носительницы заболевания или больной (при Х-сцепленном доминантном заболевании) составляет 50%
В последние годы понимание механизмов Х-сцепленной умственной отсталости дополнилось знаниями о роли Х-инактива- ции в ее генезе Х-инактивация представляет собой механизм, за счет которого достигается подавление функциональной активности в клетках у женщин одной из двух хромосом X В норме Х-инактивация приводит к тому, что в каждой клетке женского организма активна только одна из двух хромосом X, полученная женщиной от матери или от отца (то есть имеет место своего рода функциональный мозаицизм) Как правило, от отцовской и материнской хромосом X в организме женщины функциональный вклад составляет по 50% Однако, если женщина имеет мутацию Х-сцепленного гена, тогда может нарушаться рост той группы клеток, где активна хромосома X, несущая мутацию Следовательно, происходит направленная селекция клеток в пользу тех, где активна непораженная хромосома X В результате возникает «сдвиг Х-инактива- ции», вплоть до полного отсутствия клеток с активной пораженной хромосомой X
Наглядным примером может служить Х-сцепленная субкортикальная ламинарная гетеротопия (или синдром «двойной коры») Мутации ответственного за патологию гена ведут к нарушению миграции нейронов Благодаря X инактивации, у больных женщин одновременно сосуществуют две группы нервных клеток одна - с активной здоровой хромосомой X формирует нормальную кору, а другая - с активной хромосомой X, несущей мутацию, отстает в процессе миграции, формируя второй слой коры - массивное скопление серого вещества в виде ленты между корой большого мозга и желудочками, которое выявляется при магнитно- резонансной томографии
Сдвиг Х-инактивации является характерным признаком многих Х-сцепленных болезней Так, в общей популяции он встречается не более чем у 10% женщин, а в семьях, где есть больные с различными Х-сцепленными заболеваниями, - у половины всех женщин [1 ]
Исследования Х-инактивации открывают новые возможности для профилактики Х-сцепленных болезней Сдвиг Х-инактива- ции может привести как к очень тяжелому течению болезни, так и к стертым формам и даже в случае 100% сдвига - к бессимптомному носительству [2] Иными словами, сдвиг Х-инактивации у клинически здоровой женщины может быть признаком бессимптомного носительства ею Х-сцепленно- го генетического дефекта У этих женщин- носительниц риск рождения больного ребенка равен 50%, поэтому их выявление очень важно для медико-генетического консультирования
В данном разделе представлены наиболее заметные в педиатрической практике Х-сцепленные формы наследственных заболеваний
4.3.1. Х-сцепленная форма умственной отсталости
Х-сцепленная умственная отсталость
(XLMR - X-linked mental retardation) представляет собой клинически гетерогенную группу наследственных заболеваний, обусловленных мутациями генов хромосомы X и приводящих к нарушению развития когнитивных способностей
Связь умственного дефекта с половыми различиями отмечалась исследователями с конца XIX века в специализированных учреждениях больных с умственной отсталостью мужчин было на 40% больше, чем женщин Позднее этот факт объяснили высокой распространенностью мутаций Х-сцеленных генов, эффект которых ярко проявляется именно у мальчиков, поскольку они имеют единственную хромосому X Эта концепция была подтверждена последующими клиническими и молекулярными исследованиями Эпидемиология. Частота Х-сцепленной умственной отсталости чрезвычайно велика, сравнима с частотой синдрома Дауна и достигает 1 на 550-600 мальчиков В долевом отношении это свыше '/5 когнитивных расстройств у мужчин или около половины всех наследственных форм умственной отсталости
Классификация. В настоящее время известно более 200 форм Х-сцепленной умственной отсталости На основании клинических проявлений их разделяют на сицдро- мальные и несиндромальные При синдро- мальных формах умственная отсталость сочетается с характерными соматическими, неврологическими или метаболическими аномалиями Несиндромальные формы проявляются только умственной отсталостью Их диагностика очень сложна в связи с недостатком отличительных клинических критериев, за исключением снижения интеллекта у пробанда и Х-сцепленного характера наследования патологии Однако в спорадических случаях умственной отсталости генеалогический анализ не дает информации о типе наследования Поэтому значительная часть Х-сцепленных несиндромальных форм не распознается, оставаясь в группе детей с недифференцированной умственной отсталостью
Синдромальную XLMR подразделяют на четыре группы
1) наиболее многочисленная (79 нозоло- гий) - это синдромы с мальформациями, их объединяет наличие врожденных ано малий органов и систем (например, синдром умственной отсталости, сцепленной с ломкой хромосомой X (Fragile X-syn- drome, FRAXA), Х-сцепленная гидроцефалия, синдром Коффина-Лоури и др),
2) нервно-мышечные болезни представлены 37 нозологиями, наиболее частой формой является мышечная дистрофия Дюшенна,
3) метаболические болезни выделяются отдельно, поскольку в их основе лежат нарушения функций специфических энзимов - всего описано 12 состояний, таких как адренолейкодистрофия, мукополисахаридоз II типа, синдром Леша-Нихана, синдром Менкеса и др,
4) доминантные состояния в этой классификации помещены обособленно из-за их специфического наследования, при котором больны только девочки, а пораженные мальчики отсутствуют, так как, за единичными исключениями, погибают еще внутриутробно К доминантным состояниям относят 8 нозологических форм (например, синдром Блоха-Сульцбергера, синдром Ретта и др)
|
|
|
