 |


Дифференциация популяций по грузу наследственных болезней 31 глава
|
|
|
|
В отличие от менделевских (моногенных) болезней, предсказание на основе генетической информации будет ли подвержен тот или иной индивидуум мультифак- ториальной болезни, представляет сложную проблему, поскольку в этом случае наличие генетического дефекта не эквивалентно наличию болезни Более того, в случае мультифакториальных болезней не ожидается, что мутационные дефекты каких-либо отдельных генов послужат причиной для всех или большинства случаев болезни Исходя из этого, можно заключить, что генетическая расшифровка мультифакториальных болезней является сложной задачей и, вероятно, будет представлять
Литература
1 Бочков Н П Теоретические и организационные основы профилактики наследственных болезней Профилактика наследственных болезней М, 1987, 5-16
2 Gelehrter Т D, Collins F S, David Ginsburg Principles of Medical Genetics Baltimore Williams and Wilkins, 1998
3 Bell J I Polygenic disease Curr Opin Genet Devel 1993,466-9
4 Carter С 0 Multifactorial genetic disease Hosp Pract 1970 5 45-59
5 Keats В Population genetics In D L Rimoin, J M Connor, R E Pyeritz, eds Emery and Rimoin's principls and practice of medical genetics 3" ed N Y Churchill Livingstone University, 1996,347-57
главную проблему медицинской генетики в ближайшем будущем.
Успехи, достигнутые в процессе реализации программы «Геном человека», выделение и расшифровка молекулярной организации генов, изучение причин их патологии несомненно будут способствовать разработке профилактических мероприятий и выявлению групп людей, склонных к мультифакториальным заболеваниям
Клинические проявления отдельных форм мультифакториальных болезней у детей представлены в соответствующих разделах руководства (кардиология, гастроэнтерология, нефрология и др)
6 Бочков Н П Клиническая генетика М ГЭОТАР- Мед, 2001,448
7 Пузырев В П, Карпов Р С, Степанов В А, Салю- ков В Б Скрининг генов подверженности к атеросклерозу 1-я Всерос конф по проблемам атеросклероза, посвященная 100-летию со дня рождения А Л Мясникова 8-9 июня 1999 г М, 1999, 49
|
|
|
8 Леонтьева И В Атеросклероз, ишемическая болезнь сердца, инфаркт миокарда как педиатрические проблемы Клин лекция Российский вестник перинатологии и педиатрии 1997, (прил) 58
9 Бочков Н П, Захаров А Ф, Иванов В И Медицинская генетика М Медицина, 1984, 368
4.6. Болезни накопления и болезни клеточных органелл
4.6.1. Мукополисахаридозы
Среди всех болезней накопления в педиатрической практике с наибольшей частотой встречаются мукополисахаридозы При этой патологии в результате недостаточности ли- зосомальных ферментов изменяется катаболизм основного вещества соединительной ткани - гликозаминогликанов, происходит накопление их в лизосомах, что приводит к грубой клеточной патологии и возникновению характерной клинической картины.
Первые сведения о мукополисахаридо- зах появились в 1917 г, когда С Hunter описал у двух братьев 8 и 10 лет изменения опорно-двигательного аппарата, гепа- тосплено- и кардиомегалию, а также небольшое снижение интеллекта. Два года спустя G Gurler наблюдала идентичную, но более тяжелую клиническую картину заболевания у двух мальчиков, не состоявших в кровном родстве.
Впоследствии внешнее сходство больных с фигурами, украшавшими собор Парижской Богоматери, послужило основанием для объединения данной группы заболеваний под названием «гаргоилизм».
В 1952 г. G.Brante выделил из печени больных фракцию, содержавшую гексоза- мин и уроновую кислоту. Именно с этого года, благодаря данным исследованиям, в литературе появилось новое современное название патологии - «мукополисахари- дозы». Дальнейший анализ показал, что выделенные вещества являлись гликоза- миногликанами, затем были идентифицированы их количественный и фракционный составы и определены показатели почечной экскреции.
|
|
|
В зависимости от ферментативных дефектов и тяжести клинической симптоматики в настоящее время выделяют 14 типов мукополисахаридозов[1]
| Классификация мукополисахаридозов | |
| ІНтип | синдром Гурлер |
| мукополисахаридоза | |
| IH/S тип (I-V тип) | синдром Гурлер-Шейе |
| IS тип | синдром Шейе |
| II тип | синдром Хантера, |
| мукополисахаридоза | легкая и тяжелая формы |
| III тип | синдром Санфилиппо |
| мукополисахаридоза | |
| IIIA тип | синдром Санфилиппо А |
| IIIB тип | синдром Санфилиппо В |
| НІС тип | синдром Санфилиппо С |
| IIID тип | синдром Санфилиппо D |
| IV тип | синдром Моркио |
| мукополисахаридоза | |
| IVA тип | синдром Моркио А |
| IVB тип | синдром Моркио В |
| VI тип | синдром Марото-Лами, |
| мукополисахаридоза | легкая и тяжелая формы |
| VII тип | синдром Слая |
| мукополисахаридоза |
В практическом здравоохранении все типы мукополисахаридозов удобнее делить на две группы - «гурлер-подобный» и «моркио-подобный» фенотипы. Последний включает синдромы Моркио А и В, а остальные 12 объединяет «гурлер-подоб- ный» фенотип.
Внешние признаки различных типов мукополисахаридозов довольно специфичны. Они проявляются в задержке роста, диспропорциональном строении скелета (короткие туловище и шея, длинные конечности), в грубых чертах лица, костных деформациях (грудины, черепа, позвоночника, конечностей), в тугоподвижности крупных и мелких суставов При этом, как правило, отмечаются редкие зубы, дистрофия зубной эмали, множественный кариес, макроглоссия, гипертелоризм глаз, запавшее переносье, низкорасположенные ушные раковины, гепатоспленомега- лия, пахово-мошоночные и пупочные грыжи, гипертрофия лимфоидного глоточного кольца. Типичны патологии со стороны ЦНС (снижение интеллекта, часто довольно грубое), органа зрения (помутнение роговицы, глаукома), сердечно-сосудистой системы (чаще недостаточность клапанов сердца) и слуха (тугоухость).
4.6.1.1. Мукополисахаридоз IH типа (синдром Гурлер)
IH тип мукополисахаридоза - синдром Гурлер встречается с популяционной частотой 1: 40 000-1: 100 000.
Тип наследования - аутосомно-рецессивный.
Гэнетические данные и патогенез. Ген
I типа мукополисахаридоза локализован в области длинного плеча хромосомы 22, в локусе 22q 11 [1]. Патология обусловлена гетерогенной группой мутаций в гене, кодирующем лизосомный фермент a-L-идуро- нидазу В настоящее время продолжает разрабатываться молекулярная диагностика синдрома Гурлер. В нашей стране ДНК- диагностика мукополисахаридозов осуществляется в Медико-генетическом научном центре РАМН, в лаборатории, возглавляемой Е.Ю.Захаровой.
|
|
|
Наиболее частыми мутациями у европейцев считаются Q70X и W402X, в то время как мутация R89Q наблюдается преимущественно у японцев
В российской популяции соотношение распространенности аллелей W402X и Q70X составляет 4 и 44%, что наиболее близко к аналогичным показателям среди населения скандинавских стран (17% и 62%, соответственно) и значительно отличается от таковых у англо-саксонских жителей
В нашей стране 50% носителей аллеля Q70X находятся в Центрально-Европейском регионе России (Московская, Владимирская и Тульская области) Наряду с этим установлено, что у больных других этнических групп бывшего СССР (татары, грузины, армяне, узбеки, туркмены) развитие заболевания ни в одном случае не было обусловлено мутациями W402X и Q70X [2]
Сравнительный анализ показал, что го- мозиготность или смешанная гомозигот ность по мутациям W402X и Q70X влечет за собой формирование тяжелой клинической симптоматики синдрома Гурлер, в то время как мутация R89Q сопровождается более легким течением болезни [3]
Патоморфологические изменения формируются за счет отложения гликозами- ногликанов в органах и тканях больных
Клиническая характеристика. Синдрому Гурлер свойственны наиболее тяжелая клиническая симптоматика и ранняя манифестация болезни с поражением ведущих органов и систем При I типе му- кополисахаридоза наблюдаются грубые черты лица, гирсутизм, низкий рост, патология опорно-двигательного аппарата (тугоподвижность крупных и мелких суставов, кифозы, кифосколиозы), диффузная мышечная гипотония, грыжи, гепато- и спленомегалия, изменения сердечнососудистой (гипертрофическая кардио- миопатия, недостаточность клапанов) и центральной нервной систем (снижение интеллекта, обычно довольно грубое) Для детей с синдромом Гурлер типичны также поражения органов зрения (помутнение роговицы, глаукома) и слуха (тугоухость) (рис 4 6 1)
|
|
|
Лабораторные и инструментальные исследования. Диагноз IH типа мукополи- сахаридоза ставится на основании клини- ко-лабораторных и рентгено-функциональ- ных показателей
В лейкоцитах и лимфоцитах крови, биоптатах печеночной ткани и культуре кожных фибробластов определяется крайне низкая (вплоть до нулевой) активность лизосомного фермента ce-L-идуро- нидазы
Наряду с этим отмечается высокая (в 5-10 раз превышающая норму) почечная экскреция гликозаминогликанов и, как правило, низкая - оксипролина Фракционный состав гликозаминогликанов мочи представлен преимущественно гепаран- и дерматансульфатами
На ЭКГ обычно обнаруживают признаки гипертрофии миокарда левого желудочка, неспецифические изменения процесса ре- поляризации, удлинение интервалов P-R и Q-T
Рентгенографические исследования констатируют уменьшение высоты тел позвонков, укорочение и утолщение их отро-

Рис 461 Ребенок с синдромом Гурлер (I тнл муконо- нисахарндоза).

стков, деформацию грудной клетки с сужением межреберных промежутков, короткие ключицы, скошенность крыши вертлужных впадин, маленькие, уплощенные головки бедренных костей, расширение диафизов, истончение кортикального слоя, отставание костного возраста, краниостеноз, недоразвитие верхней челюсти и носовых костей, склероз сосцевидных отростков, кар- диомегалию, иногда кальцификацию кольца митрального клапана [3] При ультразвуковом исследовании диагностируют узловое утолщение створок клапанов сердца и укорочение хорд Электронно-микроскопические исследования клеток различных органов и тканей выявляют крупные вакуоли, ограниченные одноконтурной мембраной и содержащие большие количества гликоза- миногликанов.
Дифференциальный диагноз проводится с другими типами мукополисахаридозов, обусловленных дефектом фермента a-L-идуронидазы. I-V и V типы болезни (синдромы Гурлер-Шейе и Шейе) характеризуют более легкая симптоматика и незначительно сниженный или нормальный интеллект (рис. 4.6.2; 4 6.3).
4.6.1.2. Мукополисахаридоз II типа (синдром Хантера)
Частота в популяции составляет 1: 70 000-1: 200 ООО.
Тип наследования - рецессивный, сцепленный с Х-хромосомой.
Генетические данные и патогенез. Ген картирован на длинном плече Х-хромосомы, в локусе Xq27.1-q28.
В основе болезни лежит отсутствие активности лизосомного фермента идуронат- сульфатазы, принимающего участие в катаболизме гликозаминогликанов преимущественно гепаран- и дерматансульфатов.
|
|
|

Рис 4 62 Ребенок с 1-У типом мукополисахаридоза (сиидром Гурлер-Шейе).

Рис 4 63 Ребенок с V типом мукополисахаридоза (синдром Шейе).
Окончательная идентификация спектра мутаций гена идуронатсульфатазы к настоящему времени еще не завершена. Тяжелую форму синдрома Хантера связывают с мутацией L279X, открытой в 1996 г. [4].
Синдромом Хантера страдают, как правило, только мальчики. Однако в 1977 г. появилось сообщение о двух девочках с характерными проявлениями мукополиса- харидоза II типа [5]. У одной из больных брат также страдал идентичным заболеванием, а родители были жителями маленького городка В другой семье родители оказались родственниками (троюродные брат и сестра). По всем изученным параметрам (накопление меченых гликозаминогликанов, активность идуронат- сульфатазы в лимфоцитах и гомогенатах тканей) никаких отличий от классического синдрома Хантера не выявлено. Анализ аналогичных показателей у родителей не зарегистрировал отклонений. Клоны у обеих матерей также были нормальными. Результаты проведенных исследований позволили сделать предположение о существовании редкого аутосомно-рецес- сивного гена, контролирующего активность фермента идуронатсульфатазы. 14 лет спустя было опубликовано еще одно наблюдение с описанием девочки с клиническим симптомокомплексом синдрома Хантера [6] Снижение активности лизо- сомного фермента идуронатсульфатазы в лимфоцитах больной подтвердило наличие у нее мукополисахаридоза II типа Дальнейшие исследования лимфоцитов периферической крови ребенка обнаружили инактивацию полученной от матери Х-хромосомы, несущей нормальный ген идуронатсульфатазы. Инактивация Х-хро- мосомы носила, по мнению авторов, случайный характер. В результате данного феномена проявилась мутация гена идуронатсульфатазы, полученного девочкой от своего отца с синдромом Хантера. Следующее сообщение о мукополисахаридо- зе II типа у девочки появилось в 1992 г. [7]. В этом сообщении описана больная из монозиготной близнецовой пары, что дало основание исследователям высказать гипотезу о наличии ассоциации между факторами многоплодия и избирательной инактивацией Х-хромосомы.

Рис 464 Узелкеее-папулезяее поражение кежи у больного с синдремем Хантера (II тип мукенелисл- харидеза).
Клиническая характеристика. Синдром Хантера отличается от мукополисахаридоза I типа (синдрома Гурлер) более поздней манифестацией (на первом году жизни), менее выраженной умственной отсталостью, отсутствием помутнения роговицы и более благоприятным прогнозом.
Для данной формы мукополисахаридоза характерно узелково-папулезное поражение кожи преимущественно в области лопаток, наружных и боковых поверхностей плеч и бедер (рис. 4.6.4). Эти изменения обусловлены отложением липидов и гликозаминогликанов в дерме. При синдроме Хантера сердце также вовлекается в патологический процесс, при этом чаще регистрируется патология митрального клапана.
Синдром Хантера по тяжести клинической симптоматики делится на две формы (тяжелую и легкую). У больных с тяжелой формой течения наблюдаются выраженная умственная отсталость и меньшая продолжительность жизни (как правило, не превышающая 15 лет). Дети с более легкой формой заболевания отличаются нормальным интеллектом и большей продолжительностью жизни (до 40 и более лет). Они могут обучаться по общеобразовательной программе, успешно заканчивать высшие учебные заведения и не менее успешно работать по специальности.
Лабораторные и инструментальные исследования. Диагноз синдрома Ханте- pa ставится на основании клинико-генеа- логических данных наличие в родословной больных мужского пола по материнской линии, а также мужской пол больного ребенка, манифестация заболевания на первом году жизни, отсутствие помутнения роговицы, гурлер-подобный фенотип про- гредиентное течение болезни
У больных II типом мукополисахаридоза определяются крайне низкие показатели активности лизосомного фермента идуро- натсульфатазы в лейкоцитах, культуре фибробластов кожи и биоптатах печени От мечается высокая экскреция с мочой суль фатированных фракций гликозаминогли канов - гепаран- и дерматансульфатов
На ЭКГ у детей с синдромом Хантера регистрируется снижение вольтажа желудочковых комплексов QRS
Рентгенологические данные напомина ют изменения при мукополисахаридозе I типа, но они менее выражены Одним из дифференциально-диагностических кри териев между синдромами Гурлер и Хантера являются изменения кистей, при этом больным с мукополисахаридозом II типа свойственны лишь небольшие сужения проксимальных отделов пястных костей и гипоплазия ногтевых фаланг
Патоморфологические изменения однотипны с синдромом Гурлер Морфологический анализ мио и эндокарда выявляет небольшие суданофильные клетки, запол ненные гликолипидами Створки митраль ного клапана утолщены и регидны
4.6.1.3. Мукополисахаридоз III типа (синдром Санфилиппо)
Синдром Санфилиппо, или III тип муко полисахаридоза, был впервые описан JSSanfilippo et al в 1963 г Это одна из наиболее тяжелых форм патологии Выделяют четыре типа заболевания А, В, С и D Для каждого типа характерна недостаточность своего лизосомного фермента
Суммарная частота синдрома Санфи липпо в популяции составляет 1 30 ООО
Тип А наиболее часто встречается в Ни дерландах (1 24 ООО) В других популяци ях его частота не поевышает 1 200 ООО [1 ] Данные о частоте синдрома Санфилип по В довольно противоречивы Ряд авторов считает, что тип В чаще регистрирует ся на юге Европы а в других регионах наблюдается с такой же частотой как синдром Санфилиппо А (1 200 000)
Мукополисахаридоз IIIC типа регистри руется в 5-6 раз реже чем тип В
Первые сообщения о синдроме Санфилиппо D сделаны L С Ginsburg et al, R Mata- lon et al, соответственно в 1977 и 1978 гг Исследователи ошибочно расценили дан ные наблюдения как VIII тип мукополисаха ридоза В 1980 г Н Kresse установил, что описанные ранее эти два случая соответствовали синдрому Санфилиппо D
Тип D встречается крайне редко его частота не установлена В настоящее время в мировой литературе описано всего десять случаев заболевания у четырех этни ческих групп - выходцев из Восточной Индии, Сардинии, Италии и Северной Америки В нашей стране сведения о синдроме Санфилиппо D отсутствуют
Генетические данные и патогенез. Все типы синдрома Санфилиппо наследу ются аутосомно-рецессивно
Синдром Санфилиппо А обусловлен от сутствием активности лизосомной гидро лазы гепаран N сульфатазы Ген патоло гии картирован на длинном плече хромосомы 17, в локусе q 25 3
Синдром Санфилиппо В связан с край не низкой активностью лизосомного фер мента М-ацетил-N-a D глюкозаминидазы Ген локализован также на длинном плече хромосомы 17, в районе q 21
В основе синдрома Санфилиппо С нахо дится дефицит фермента ацетил-КоА а-глюкозаминид-М-ацетилтрансферазы Локализация гена определяется на хромо соме 14 Более точное картирование гена пока не проведено
При синдроме Санфилиппо D резко снижена активность лизосомной гидрола зы N-ацетилглюкозамин-б-сульфатсуль- фатазы Ген патологии картирован в области длинного плеча хромосомы 12, в локусе q14
В результате крайне низкой активности перечисленных лизосомных ферментов нарушается катаболизм и происходит отложение в клетках органов и тканей больных одного из сульфатированных гликозаминогликанов - гепарансульфата. Наряду с этим отмечается высокая почечная экскреция этого сульфатированного метаболита соединительной ткани
Клиническая характеристика. Большинство исследователей считают, что нет четких дифференциально-диагностических клинических критериев между различными типами синдрома Санфилиппо
Заболевание развивается обычно на 2 году жизни Болезнь носит прогреди- ентный характер Анализ патолого-анато- мического материала от двух недоношенных детей с синдромом Санфилиппо А, умерших сразу после рождения, не выявил никаких включений в клетках мозга, костей и суставов новорожденных Однако был обнаружен дефицит фермента в тканях [9] III типу мукополисахаридоза свойственны, как правило, нормальные показатели физического развития, минимальные изменения скелета, небольшое увеличение паренхиматозных органов и грубое снижение интеллекта Орган зрения и сердечно-сосудистая система не всегда вовлекаются в патологический процесс Наряду с этим известны случаи тяжелой митральной недостаточности при типе В синдрома, потребовавшие хирургического вмешательства у детей 3-6 лет [10]. Клинический полиморфизм синдрома Санфилиппо В был отмечен в семье, где у двух сестер наблюдалась тяжелая форма заболевания глубокая умственная отсталость, гепато- и спленомега- лия, гирсутизм Брат девочек также страдал IIIB типом мукополисахаридоза Однако течение болезни у него было легким, с незначительным снижением интеллекта [11 ] Такая вариабельность феноти- пических проявлений внутри семьи может быть объяснена как влиянием средо- вых факторов или модифицирующих генов, так и наличием у родителей одного нормального аллеля из трех вариантов мутантных, комбинация которых обусловливает различную тяжесть поражения Не исключается также возможность мозаи- цизма, когда фермент присутствует в части клеток, предопределяя легкое течение болезни
Синдром Санфилиппо С считается одной из наиболее легких форм среди всех его типов Установлено, что заболеванию свойственны незначительные изменения скелета, минимальное увеличение паренхиматозных органов и небольшое снижение интеллекта [12]
Малое количество описанных больных не дает четкого представления о характере клинической симптоматики синдрома Санфилиппо D Проявления заболевания, как и при других типах, начинаются обычно на втором-третьем годах жизни. Наряду с этим описана больная с задержкой раннего психомоторного развития вставать стала с одного года, ходить - с двух лет, говорить - с двух с половиной лет Однако запас слов ограничивался только несколькими фразами. В четыре года стал заметен регресс приобретенных навыков, появились признаки агрессии В десять лет ребенок перестал самостоятельно ходить, говорить и не мог обходиться без посторонней помощи
Для синдрома Санфилиппо D типично отсутствие или незначительные проявления тугоподвижности суставов. Характерна гипоплазия зубовидного отростка Сердечно-сосудистая система и паренхиматозные органы обычно не вовлекаются в патологический процесс или их поражение выражено минимально. Большинство исследователей отмечают наличие лицевых «гурлер-подобных» аномалий. Описан также гирсутизм Снижение интеллекта свойственно всем больным, но степень задержки психоречевого развития варьирует и колеблется от легкой степени до идиотии Описана девочка, посещавшая в течение пяти лет начальную школу и, как отмечают исследователи, практически без успеха У одного ребенка диагностирована хроническая диарея, однако единичное наблюдение не дает основания считать поражение желудочно-кишечного тракта патогномоничным для синдрома Санфилиппо D.
Диагностика. Диагноз синдрома Санфилиппо ставится на основании клинической симптоматики и результатов лабораторных методов исследования Отмечается снижение активности соответствующих лизосомных гидролаз в лейкоцитах, культуре фибробластов кожи и биоптатах печеночной ткани. Характерно повышение почечной экскреции гликозаминогликанов. Фракции ГАГ мочи представлены в основном гепарансульфатом
4.6.1.4. IV тип мукополисахаридоза (синдром Моркио)
Синдром Моркио, или IV тип мукополисахаридоза, был впервые описан в 1929 г уругвайским педиатром L Morquio и J.F Brailsfort в Англии Выделяют два типа этого заболевания - синдромы Моркио А и В
По данным исследователей, частота синдрома Моркио А в популяции колеблется от 1 40 ООО до 1. 300 ООО, а частота Моркио В - 1.300 000
Генетические данные и патогенез. Болезнь наследуется по аутосомно-рецессив- ному типу Ген синдрома Моркио А картирован на длинном плече хромосомы 16, в локусе q 24 3-16q 24 3 Локализация гена синдрома Моркио В пока не установлена
В основе болезни лежит генетически детерминированное отсутствие активности ферментов N-ацетилгалактозамин-б-суль- фатсульфатазы (Моркио А) и р-галактози- дазы (Моркио В), в результате чего нарушается катаболизм сульфатированного гликозаминогликана основного вещества соединительной ткани - кератансульфата и происходит его накопление в клетках органов и тканей больных
Клиническая характеристика. Синдрому Моркио А и В свойственны диспропорциональная карликовость, типичные изменения внешности грубые черты лица, большой рот, прогнатизм Больным свойственны также килевидная деформация грудной клетки, увеличение суставов с их минимальной тугоподвижностью или нормальным объемом движений, укорочение туловища, кифоз, кифосколиоз, поясничный лордоз, вальгусная деформация нижних конечностей, аномалии зубов (рис 4.6 5) Наряду с этим к характерным признакам относятся диффузная мышечная гипотония, грыжи, гепато- и сплено- мегалия, недостаточность клапанов сердца, снижение зрения вследствие помутнения роговицы, тугоухость Ю (коэффициент интеллектуального развития), как правило, нормальный и составляет 90-115 единиц
Наиболее тяжелым проявлением болезни считают нарушение атланто-акципи- тального сочленения, которое может приводить к острой компрессии спинного мозга и внезапной смерти ребенка
Особенностями синдрома Моркио В являются более поздняя манифестация заболевания и меньшая степень тяжести болезни
Диагноз IV типа мукополисахаридоза ставится на основании клинической симптоматики, результатов рентгенологического исследования, крайне низкой активности лизосомных гидролаз - N-ацетилга- лактозамин-6-сульфатсульфатазы (синдром Моркио А) и р-галактозидазы (синдром Моркио В) в лейкоцитах крови, а также высокой почечной экскреции кератансульфата Рентгенологически выявляются платиспондилия, искривление дистальных частей локтевой и лучевой костей, деформации эпифизов трубчатых костей, расширение метафизов, ребер, короткие фаланги, деформация метакарпальных костей, остеопороз
Синдромы Моркио А и В необходимо дифференцировать с рахитоподобными заболеваниями - почечным тубулярным ацидозом и болезнью де Тони-Дебре-Фан- кони Нормальные показатели минерального обмена сыворотки крови и мочи (активность щелочной фосфатазы, уровень кальция, фосфора в сыворотке крови и величины их почечной экскреции), большие количества выводимых с мочой гликозаминогликанов, наличие помутнения роговицы, тугоухости, грубых черт лица позволяют поставить правильный диагноз IV типа мукополисахаридоза, или синдрома Моркио Исследование активности лизосом- ных гидролаз лимфоцитов помогает разграничить тилы А и В
4.6.1.5. VI тип мукополисахаридоза (синдром Марото-Лами)
Первые два случая синдрома Марото- Лами были описаны Н R Taylor et al в 1978 г [13] Авторы описали двух больных из семьи австралийских аборигенов с характерными внешними признаками синдрома Гурлер, но с нормальным уровнем в лимфоцитах фермента a-L-идуронидазы и пониженной активностью арилсульфатазы В Родители этих детей были кузенами Частота заболевания не установлена Генетические данные и патогенез. Патология наследуется по аутосомно-ре- цессивному типу
В основе болезни лежит дефицит лизо- сомного фермента арилсульфатазы В, принимающего участие в катаболизме сульфатированной фракции гликозаминогликанов - дерматансульфата
Клиническая симптоматика синдрома Марото-Лами проявляется в карликовом росте больных с выраженными изменениями опорно-двигательного аппарата, патологии сердечно сосудистой системы и других па ренхиматозных органов, поражении органов зрения и слуха при сохраненном (нормальном) интеллекте Выделяют две формы болезни - легкую и тяжелую (рис 4 6 6, 4 6 7)
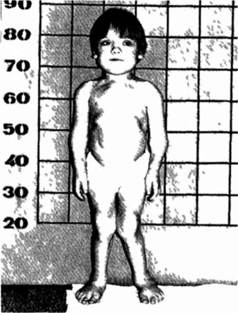
Рис 465 Ребенок с синдромом Моркио A (IVA ТИП мукополисахаридоза).
Диагноз VI типа мукополисахаридоза ставится на основании генеалогических данных, совокупности клинических признаков, крайне низкой активности лизо- сомного фермента арилсульфатазы В в лейкоцитах крови и экскреции с мочой дерматансульфата
4.6.1.6. VII тип мукополисахаридоза (синдром Слая)
Заболевание впервые описано W S Sly, В A Quinton, et аі в 1973 г
Мукополисахаридоз VII типа встречается крайне редко Частота его не установлена Синдром Слая наследуется по ауто- сомно-рецессивному типу Патология обусловлена дефицитом фермента р-глюкуро- нидазы
Ген синдрома Слая картирован на хромосоме 7, в локусе 7q21 11
В результате проведенного молекулярного анализа у больных из 17 семей с VII типом мукополисахаридоза идентифицировано 20 различных мутаций в гене р-глюкуронидазы, из них 14 мутаций выявлены впервые [14]
В организме больных с VII типом муко полисахаридоза накапливаются хондрои тинсульфаты
Клиническая характеристика. Бо лезнь выявляется в первые недели жизни (на 5-7 нед) Характерен «гурлер подобный» фенотип низкий рост аномальное строение черепа, грубые черты лица, короткая шея, кифосколиоз, косолапость, помутнение роговицы и в ряде случаев отсутствие гепато и спленомегалии Харак терно грубое снижение интеллекта Описаны шесть случаев крайне тяжелой не онатальной формы заболевания, сопрово ждавшихся водянкой и внутриутробной ги белью плода
Лабораторные и инструментальные исследования. При рентгенологическом исследовании выявляются множественный дизостоз, клювовидные позвонки, наруше ние процесса окостенения медиальных за пястных и предплюсневых костей
В лейкоцитах больных определяется крайне низкая активность лизосомного фермента (і-глюкуронидазьі
С мочой экскретируются большие КОЛИ чества гликозаминогликанов, фракции ко торых представлены преимущественно хондроитинсульфатами
Дифференциальный диагноз мукополисахаридозов с «гурлер-подобным» фенотипом следует проводить, прежде всего, с другими болезнями накопления маннози дозом, ганглиозидозом Gm 1 (болезнь Нормана-Ландинга), сиалидозом II типа, муколипидозами
|
|
|
