 |


Этапы функционирования нейтрофилов как клеточных эффекторов острого воспаления 5 страница
|
|
|
|
Интенсивность синтеза жирных кислот как этапа образования жира в виде триглицеридов, зависит не только от размеров адипоцитов, но и от соотношения на уровне всего организма секреции и эффектов главного анаболического гормона инсулина и его гормонов-антагонистов. Превалирование активности инсулина усиливает липогенез.
Интенсивность липогенеза меньше, когда человек ест часто и немного, и, наоборот, она больше, когда он ест много и редко.
Ожирение служит причиной ряда сдвигов эндокринной регуляции обмена веществ и представляет собой следствие эндокринопатий. Эндокри- нопатии при этом могут быть следствием избыточного потребления источников свободной энергии с пищей. Наиболее частое из нарушений эндокринной регуляции обмена веществ у больных с ожирением - это повышенная активность в крови инсулина, гиперинсулинемия. Чем больше ожирение, тем больше концентрация инсулина в крови утром и натощак. Гиперинсулинемия у части больных с ожирением приводит к большей суммарной длительности действия гипогликемии как внутреннего стимула к потреблению пищи. Кроме того, у больных с ожирением усилена реакция роста секреции инсулина в ответ на действие ее обычных стимулов (гипер- гликемия и др. ).
У части больных с ожирением выявляют сниженную секрецию в кровь гормона роста в ответ на ее обычные стимулы:
♦ вызванное инсулином перемещение глюкозы в клетки, вызывающее гипогликемию;
♦ рост концентрации в циркулирующей крови аргинина после внутривенной инфузии раствора аминокислоты.
Как у мужчин, так и у женщин, страдающих от ожирения, в крови снижается содержание тестостерона и эстрадиола. Концентрация же эстрона растет. Это происходит вследствие усиления его образования в строме жировой ткани из андрогенного кортикостероида. Из-за роста содержания в циркулирующей крови эстрона ожирение у женщин считают фактором риска злокачественных опухолей матки.
|
|
|
Избыточное потребление с пищей потенциальных источников свободной энергии для клеток в виде жиров и углеводов приводит к ряду взаимосвязанных патологических сдвигов обмена веществ и работы функциональных систем.
Через гипергликемию избыточное поступление нутриентов во внутреннюю среду ведет к повышенной секреции инсулина. Гиперинсулинемия вызывает резистентность клеток к эффекту инсулина на рецепторном и по- стрецепторном уровнях. Кроме того, гиперинсулинемия связана с гиперплазией инсулинобразующих клеток островков Лангерганса, которая на определенном этапе своего развития может служить причиной недостаточности внешнесекреторной функции поджелудочной железы (схема 11. 1).
Гиперинсулинемия повышает утилизацию аминокислот как субстратов белкового синтеза на уровне всего организма. Это в частности вызывает гиперплазию гладкомышечных элементов стенки сосудов сопротивления. В результате их просвет сужается, общее периферическое сосудистое сопротивление растет и возникает артериальная гипертензия. Избыточное потребление источников свободной энергии с пищей активирует симпатический отдел автономной нервной системы и повышает секрецию щитовидной железой триийодтиронина, что вызывает рост потребления кислорода всем организмом. Вслед за ростом потребления кислорода повышается минутный объем кровообращения, что вызывает артериальную гипертензию и (или) предрасполагает к ней (схема 11. 2).
|
|

Схема 11. 1. Ожирение как причина и фактор риска неинсулинзависимого сахарного диабета
|
|

Схема 11. 2. Ожирение как фактор риска и причина артериальной гипертензии
|
|
|
Избыточно^ потребдеиие. с пищей источников свободной энергии ведет к гиперлипиде9«ии и аккумуляции свободной энергии в виде триглицеридов жировой ткани. Это повышает интенсивность обмена холестерина на системном уровне. В результате растет экскреция холестерина с желчью, что ведет к образованию камней в просвете желчного пузыря. Поэтому ожирение считают фактором риска хронического калькулезного холецитита.
ГИПЕРЛИПИДЕМИИ/ГИПЕРЛИПОПРОТЕИНЕМИИ
Гиперлипидемия/гиперлипопротеинемия - патологическое состояние предболезни или заболевание, связанные с ростом содержания в плазме крови свободных жирных кислот, триглицеридов, холестерина, хиломикронов и липопротеинов (ЛП).
Существует прямая связь между концентрацией липидов в плазме крови и вероятностью атеросклероза как причины ишемической болезни сердца. Полагают, что а) гиперхолестеринемия (патологически высокая концентрация холестерина в плазме крови) и в меньшей степени рост содержания в плазме других липидов представляют собой этиологический фактор ишемической болезни сердца; б) снижение концентраций липидов через диету, физические упражнения и с использованием средств фармакологической коррекции замедляет развитие атеросклероза и ишемической болезни сердца. Эта два положения и составляют липидную гипотезу атеросклероза и ишемической болезни сердца, аргументами сторонников которой служат следующие факты:
♦ атеросклеротическая бляшка содержит липиды, большинство которых поступает в нее прямо из липопротеинов, циркулирующих с плазмой крови;
♦ атеросклеротические поражения сосудов могут быть воспроизведены в эксперименте при кормлении животных пищей с высоким содержанием холестерина;
♦ гиперлипидемию всегда выявляют у больных с окончательным диагнозом атеросклероза;
♦ результаты эпидемиологических исследований свидетельствуют о высоком риске ишемической болезни сердца как причине летальных исходов при росте в плазме крови концентрации атерогенных липопротеинов низкой плотности (ЛПНП) и снижении в ней содержания антиатерогенных липопротеинов высокой плотности (ЛПВП).
Атерогенными называют те липопротеины, рост концентрации которых в плазме крови вызывает атеросклероз, и, наоборот, неатерогенные - это ЛП, не обладающие таким свойством (табл. 11. 1).
|
|
|
Выделяют вторичную гиперхолестеринемию, то есть патологический рост концентрации холестерина в крови вследствие перемежающейся пор- фирии, блокады желчевыводящих путей, гипотиреоза и беременности. Вторичную гипертриглщеридемию (рост содержания в крови триглицеридов)
| Липопротеины и атеросклероз | Таблица 11. 1 | |
| Липопротеины | Плотность | Ате роге нность (+), |
| липопротеина | неатерогенность (0) и антиатерогенностъ (-) | |
| Лйпопротеины низкой плотности | 1, 019-1, 063 кг/л | + |
| (ЛПНП) | ||
| Липопротеины промежуточной плотнос | 1, 006-1, 019 кг/л | + |
| ти (ЛППП) | ||
| Бета-липопротеины очень низкой плот | < 1, 006 кг/л | + |
| ности (Р-ЛПОНП) | ||
| Окисленные ЛПНП (лиганды к «рецеп- | + | |
| торам-мусорщикам» наружной поверхности макрофагов сосудистой стенки) | ||
| Липопротеин(а) | + | |
| Хилом икроны | ||
| Липопротеины очень низкой плотности | 0(? ) | |
| (ЛПОНП) | ||
| Липопротеины высокой плотности | 1, 063-1 Д10 кг/л | - |
обуславливают сахарный диабет, острая алкогольная интоксикация, сепсис, индуцированный грамотрицательной инфекцией, нарушения синтеза и аккумуляции гликогена гликогена I типа, а также другие патологические состояния и болезни.
Семейная гиперлипидемия первого типа - врожденное нарушение липидного обмена, обусловленное недостаточным расщеплением хиломикро- нов и липопротеинов очень низкой плотности вследствие низкой активности катализатора их гидролиза липопротеинлипазы или недостатка активатора этого фермента, аполипопротеина С-П.
Расщепление хиломикронов с участием липопротеинлипазы происходит в плазме крови, на поверхности эндотелиальных клеток и адипоцитов. Низкая активность липопротенинлипазы ведет к снижению очищения плазмы крови от хиломикронов. Семейная гиперлипидемия первого типа, обусловленная только низкой активностью липопротеинлипазы без недостатка аполипопротеина С-П, - это врожденное расстройство липидного обмена, которое наследуется по аутосомально-рецессивному типу и обусловлено мутацией 207 остатка в пятом экзоне гена липопротеинлипазы. Его характеризуют повышенное содержание в плазме крови триглицеридов и периодические обострения хронического панкреатита, начиная с неонатального периода. Панкреатит у таких больных вызывает перерождение паренхимы поджелудочной железы, вызванное высоким содержанием в крови хиломикронов Низкий относительно нормального уровень гидролиза хиломикронов и липопротеинов очень низкой плотности в просвете капилляров, прилежащих к адипоцитам, ведет к росту концентрации хиломикронов в плазме крови. Кроме того, падает захват триглицеридов адипоцитами. Это ведет к росту их концентрации в плазме выше уровня в 4 г/л. Кроме того, рост концентрации триглицеридов в плазме крови - это следствие увеличения неферментативного расщепления в ней хиломикронов, то есть не связанного с функционированием липопротеинлипазы, а обусловленного ростом содержания хиломикронов во внутренней среде организма. Высокое содержание хиломикронов и триглицеридов в плазме крови ведет к ксантоматозу кожи, непереносимости жирной пищи и гепатоспленомегалии у больных с гипер- липидемией данного типа. Так как при гиперлипидемии I типа в плазме растет концентрация неатерогенных липопротеинов очень низкой плотности и хиломикронов, то атеросклероза у таких больных обычно не развивается. Следует заметить, что системная красная волчанка ведет к сдвигам липидного обмена почти идентичным гиперлипидемии первого типа. Лабораторные исследования плазмы крови больных с гиперлипидемией первого типа выявляют снижение липолитической активности плазмы крови, не связанной с действием гепарина как активатора липолиза.
|
|
|
Гиперлипидемия второго типа-это наследственное нарушение липидного обмена, при котором у родственников или членов одной семьи как фенотипический признак выявляют патологически высокое содержание холестерина в крови (семейная гиперхолестеринемия). Эту наследуемую по аутосомально-доминантному типу гиперлипидемию характеризуют бугорчатый ксантоматоз в области сухожилий, раннее и быстрое развитие атеросклероза, ишемической болезни сердца и инфаркта миокарда, который часто служит причиной внезапной сердечной смерти в возрасте от 20 до 50 лет. Ведущим звеном патогенеза гиперхолестеринемии, высокого содержания в плазме крови атерогенных липопротеинов низкой плотности и атеросклероза у больных с гиперлипидемией второго типа является или полное отсутствие рецепторов к липопротеинам низкой плотности на наружной клеточной поверхности или нарушения их строения и функции вследствие мутации аллелей генов RbO, Rb- и Rtio. В результате наследуемой недостаточности рецепторов к ЛПНП возникает гиперхолестеринемия, которая по механизму положительной обратной связи повышает синтез атерогенных липопротеинов низкой плотности в печени (схема 11. 3).
|
|
|
Если патогенные мутации затронули один из аллелей гена, кодирующего рецептор к липопротеинам низкой плотности (больные гетерозиготные по мутантному патогенному гену), то концентрация холестерина в плазме крови составляет 5-6 г/л. У гомозиготных (патогенный ген содержит два мутантных аллеля) больных она находится в пределах 3-4 г/л. Выделяют два вида гиперлипидемии II типа, Па и lib. При a-типе в крови увеличена концентрация холестерина при нормальных содержаниях в ней липопротеинов очень низкой плотности и триглицеридов. При b-типе концентрации в крови липопротеинов очень низкой плотности и триглицеридов повышены.
Изменения обмена веществ, обусловленные злокачественными опухолями печени, часто приводят к сдвигам содержания в крови липидов, аналогичным гиперлипидемии Па.
Гиперлипидемия третьего типа - это наследуемая по аутосомально- доминантному типу недостаточность катаболизма атерогенных липопротеинов промежуточной плотности. Как синдром гиперлипидемию третьего типа характеризуют ускоренное развитие атеросклероза и частая
|
Схема 11. 3. Патогенез семейной гиперхолестеринемии |

тромбоэмболия сосудов из системы венечной артерии, сахарный диабет, ожирение, гипотиреоз и особо выраженный ксантоматоз. При исследовании плазмы крови выявляют рост содержания в ней концентраций триглицеридов и холестерина, которые липопротеины промежуточной плотности переносят от печени на периферию. О сахарном диабете у таких больных свидетельствует сниженная толерантность по отношению к глюкозе.
Гиперлипидемия четвертого типа - это наследуемое по аутосомаль- но-доминантному типу нарушение липидного обмена, которое характеризует гипертриглицеридемия, то есть аномально высокое содержание триглицеридов в плазме крови. Это наиболее частое из нарушений липидного обмена у больных, при котором атеросклероз редко поражает периферические и венечные артерии. При гиперлипидемии четвертого типа в плазме крови повышено содержание как триглицеридов, так и липопротеинов очень низкой плотности. Отдельно выделяют приобретенную гиперлипидемию четвертого типа, которую вызывают сахарный диабет, уремия, препараты из группы глюкокортикоидов, а также бета-адреномиметики.
Гиперлипидемия/гиперлипопротеинемия пятого типа - это полиэтио- логичное и варьирующее по патогенетическим механизмам нарушение липидного обмена, вследствие которого у части больных возникают ксантоматоз и панкреатит как следствия патогенно высоких концентраций в плазме крови липопротеинов очень низкой плотности и хиломикронов.
АТЕРОСКЛЕРОЗ
Атеросклероз - отложение в интиме сосудов атерогенных липопроте- инов низкой плотности (ЛПНП) вследствие взаимодействие гладкомышечных клеток стенок сосудов с атерогенными липопротеинами при их высокой концентрации в циркулирующей крови. Атеросклеротическое перерождение сосудов во многом определяет пролиферация миоцитов сосудистой стенки, индуцируемая цитокинами тромбоцитов, активированных мононук- леаров, а также агентами аутокринной регуляции (митогенами) самих гладкомышечных клеток. Термин атеросклероз описывает изменения сосудистой стенки при этом системном типовом патологическом процессе. Слово «атере» (греч. ) означает «кашица» и указывает на образование в стенках сосудов кашицеобразных липидных отложений с их дальнейшим склерозированием и уплотнением.
По определению Всемирной организации здравоохранения «Атеросклероз - это вариабельная комбинация изменений внутренней оболочки (интимы) артерий, включающая накопление липидов, сложных углеводов, фиброзной ткани, компонентов крови, кальцификацию и сопутствующие изменения средней оболочки (медии)».
При гистологическом исследовании пораженных атеросклерозом участков сосудистой стенки выявляют характерное патологическое образование, атеросклеротическую бляшку (рис. 11. 1). Ее формируют липиды, лейкоциты, гладкомышечные клетки и межклеточное вещество интимы артерий. Образованию бляшки предшествует адгезия (прилипание) циркулирующих моноцитов и лимфоцитов к артериальному эндотелию с последующим локальным накоплением пенистых клеток. Пенистые клетки представляют собой активированные мононуклеары, видоизмененные в результате интенсивного эндоцитоза липидов.
До сих пор общественное сознание связывает атеросклероз с пропитыванием холестерином сосудистой стенки, что верно лишь отчасти. Холестерин и триглицериды переносятся во внеклеточном пространстве липопротеинами. Не гиперхолестеринемия (рост концентрации холестерина выше верхнего предела нормальных колебаний) ведет к атероскдерозу, а аккумуляция в сосудистой стенке определенных липопротеинов. Некоторые ли- попротеины можно считать атерогенными, другие неатерогенными, а третьи антиатерогенными.
Под атерогенными следует понимать липопротеины, которые проникают в сосудистую стенку, где происходит их эндоцитоз макрофагами, которые в результате эндоцитоза превращаются в пенистые клетки. В последующие стадии патологического процесса происходит отложение аморфного холестерина и его кристаллов в межклеточных пространствах сосудистой стенки, то есть вне пенистых клеток. При гиперли- пидемии второго типа, которая часто приводит к атеросклерозу, пенистые клетки, насыщенные липидами, находят во внутренней оболочке сосудистой стенки.
|
|
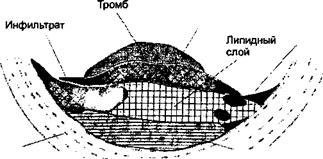
| Средняя оболочка чч кровеносных сосудов |
| Фибрин |
| Эндотелиолит |
| ^ Пролиферирующие гл адкомышеч ные клетки |
| Рис. 11. 1. Схематическое изображение строения атеросклеротической бляшки, вызывающей стенокардию |
Поверхность макрофагов содержит рецепторы к липопротеинам очень низкой плотности и непостоянные рецепторы к измененным в результате окисления липопротеинам низкой плотности («рецепторы-мусорщики»). Предположительно активация мононуклеаров через связывание этих рецепторов с окисленными атерогенными липопротеинами или липопротеинами очень низкой плотности ведет к активации макрофагов, в результате которой они приобретают способность к эндоцитозу атерогенных липопротеинов.
В ранней стадии атеросклеротического поражения сосудистой стенки в ней выявляют липидные прожилки и пенистые клетки. Этот начальный этап формирования атеросклеротической бляшки вызывает активация непостоянно присутствующих на клеточной поверхности макрофагов рецепторов к окисленным липопротеинам низкой плотности (ацетил-ЛПНП- рецепторы, «рецепторы-мусорщики»). Мусорщиками эти рецепторы называют потому, что они обладают высоким сродством к «мусору» в виде окисленных ЛПНП. Изменения ЛПНП в результате окисления, которые обуславливают их особо высокое сродство к рецепторам-мусорщикам включают:
♦ трансформацию лецитина в составе липопротеинов в лизолецитин;
♦ окисление холестерина;
♦ рост отрицательного заряда и плотности липопротеинов;
♦ снижение содержания в ЛПНП полиненасьпценных жирных кислот.
Распад структурного апопротеина липопротеинов низкой плотности В-
100 с высвобождением гистидина, лизина и пролина. Окисленные липоп- ротеины служат хемоаттрактантами и активаторами для макрофагов сосудистой стенки, одновременно являясь для них и объектом эндоцитоза.
Как результат патогенных межклеточных взаимодействий атеросклероз представляет собой патологическое изменение стенок артерий большого и среднего диаметра, которые составляют и вызывают локальная аккумуляция в интиме липидов и макрофагов, миграция и пролиферация шадкомышеч- ных клеток, а также отложения вещества внеклеточного матрикса. Патогенные межклеточные взаимодействия, приводящие к атеросклерозу, реализуются не только через действие факторов роста, но и в результате эффектов цитокинов флогогенов, медиаторов воспаления и экспрессии адгезивных молекул. Начальным этапом патогенеза атеросклероза как последовательности межклеточных взаимодействий является адгезия моноцита циркулирующей крови к эндотелиальным клеткам с их последующей миграцией в интиму. При этом индукторы атеросклероза окисленные (модифицированные) липоп- ротеины низкой плотности, воздействуя на лейкоциты циркулирующей крови и эндотелиальные клетки, вызывают экспрессию на их поверхности адгезивных молекул. Так, лизофосфатидилхолин - элемент молекулы окисленных атерогенных липопротеинов - индуцирует экспрессию межклеточной адгезивной молекулы-1 на поверхности эндотелиальных клеток. Известно, что атерогенные липопротеины оказывают на эндотелий прямые и непрямые влияния, повышающие экспрессию на их поверхности эндотелиально-лейкоцитарных адгезивных молекул (ЭЛАМ). Непрямой эффект атерогенных липопротеинов на рост экспрессии ЭЛАМ состоит в стимуляции паракринной секреции гладкомышечными клетками и мононуклеарными макрофагами сосудистой стенки. Адгезия лейкоцитов к сосудистой стенке служит первым этапом их проникновения в интиму. Там активированные моноциты секрети- руют ряд активаторов эндотелия и хемоаттрактантов, стимулирующих дальнейшую инфильтрацию очага атеросклеротического повреждения моноцитами и лимфоцитами. Патогенное функционирование иммунокомпетентных клеток в качестве эффекторов атеросклероза указывает на участие в развитии атеросклероза иммунопатологической реакции.
Атеросклероз во многом представляет собой хроническое воспаление сосудистой стенки, протекающее с преобладанием пролиферативного компонента, основными клеточными эффекторами которого являются моноциты циркулирующей крови, мононуклеарные фагоциты субинтимального слоя, гладкомышечные сосудистые клетки, активированные атерогенны- ми липопротеинами или в результате межклеточных взаимодействий.
Воспаление в очаге атеросклеротического поражения усиливает взаимосвязанное с ним локальное свертывание крови, которое могут вызвать сами атерогенные липопротеины. Так, атерогенный липопротеин(а), содержит гликопротеин(а), связанный с апопротеином В. Идентичность структуры липопротеина(а) строению плазминогена может обуславливать внутри- сосудистый тромбогенез через конкурентное связывание рецепторов к плазминогену на поверхности эндотелиоцитов.
Таков патогенез индукции атеросклероза через образование прожилок атерогенных липопротеинов и пенистых макрофагов в сосудистой стенке как начального этапа образования атеросклеротической бляшки. В нем особую роль как эффекторы и регуляторы патологического процесса играют макрофаги сосудистой стенки, которые сами могут индуцировать окисление ЛПНП через высвобождение свободных кислородных радикалов и (или) активацию липооксигеназы. Так как макрофаги представляют собой эффектор единой системы иммунитета организма, то мы вправе предположить роль в развитии атеросклероза нервного эндогенного этиологического фактора, столь сильно меняющего состояние системы иммунитета, и ее макрофа- гального звена в частности. Кроме того, патогенный стресс как состояние, которое характеризует системная интенсификация свободнорадикального окисления липидов, может повышать уровень окисления ЛПНП и на уровне сосудистой стенки предрасполагать к атеросклерозу.
Пенистые клетки высвобождают ряд цитокинов, чье действие вызывает пролиферацию клеточных элементов, и в особенности миоцитов гладкомышечных элементов сосудистой стенки. Кроме того, цитокины активированных макрофагов активируют эндотелиоцигы, что ведет к росту экспрессии их тромбогенного потенциала. Цитокины пенистых клеток активируют и нейтрофилы циркулирующей крови, что вызывает воспаление с полимор- фонуклеарами в качестве его клеточных эффекторов. В результате в составе атеросклеротической бляшки находят пролиферирующие миоциты сосудистой стенки, агрегаты активированных тромбоцитов, других форменных элементов крови, активированные нейтрофилы и нити фибрина. Все это характеризует атеросклеротическую бляшку как очаг воспаления и локус тромбоза.
Макрофаги, окисленные липопротеины, ацетил-ЛПНП-рецепторы играют определяющую роль в индукции атеросклероза. В дальнейшем процесс образования атеросклеротической бляшки теряет связь с этими этиологическими факторами, то есть через патогенные межклеточные взаимодействия происходит его эндогенизация.
Одно из наиболее впечатляющих достижений медицинской генетики последних десятилетий - выяснение генетической детерминированности гиперхолестеринемии и высокой концентрации в плазме крови липопроте- инов низкой плотности как причины атеросклероза. Исследовав 500 больных, выживших после инфаркта миокарда, Гольдштейн, Браун и Мотульс- кий более, чем в половине случаев выявили семейную гиперхолестеринемию, семейную гиперлипидемию или комбинированную гиперлипидемию. Было установлено, что эти расстройства липидного обмена представляли собой патогенные фенотипические признаки, детерминированные одним геном, то есть были моногенными наследственными расстройствами липидного обмена. Последующие исследования клеточных культур фибробластов кожи, лимфоцитов, и миоцитов аортальной стенки, взятых у гомозиготных и гетерозиготных по данному гену больных, позволили выявить генетически детерминированные дефекты связывания молекулярного комплекса липопротеины низкой плотности-холестерин с наружной клеточной мембраной и его последующего пиноцитоза.
Взаимодействие молекулярного комплекса холестерин-ЛПНП при нормальной экспрессии ЛПНП-рецептора на поверхности клеток ведет к пино- цитозу молекулярного комплекса. После пиноцитоза комплекс инкорпорируется в лизосомы, где и происходит высвобождение свободного холестерина. Рост концентрации свободного холестерина в клетке снижает активность ключевого фермента внутриклеточного синтеза холестерина гид- роксиметилппотарил-коэнзим А-редуктазы. Наследуемая по аутосомально-
доминантному типу недостаточность ЛПНП-рецепторов ведет к снижению пиноцитоза комплекса холестерин-ЛПНП и к падению концентрации свободного холестерина в клетках. В результате низкого содержания свободного холестерина в клетках с низким содержанием на наружной поверхности ЛПНП-рецепторов в них высока активность гидроксиметилглютарил-коэн- зим А-редуктазы. Это ведет к интенсивному образованию холестерина клетками, его высвобождению во внеклеточное пространство и росту в нем содержания атерогенных липопротеинов переносчиков холестерина. Гиперхолестеринемия вызывает атеросклероз не через пропитывание стенки сосудов холестерином, а через повышение интенсивности образования печенью и высвобождения ею в кровь атерогенных липопротеинов (схема 11. 3). Кроме того, гиперхолестеринемию при семейной гиперхолестери- немии и других гиперлипидемиях, связанных с атеросклерозом, обуславливает и низкий уровень связывания комплекса ЛПНП-холестерин с ЛПНП-рецепторами наружных клеточных мембран. Гиперхолестеринемия алиментарного генеза также повышает риск атеросклероза через увеличение в крови концентрации ЛПНП и других атерогенных липопротеинов, переносящих холестерин во внеклеточном пространстве. Рост концентрации ЛПНП повышает массу циркулирующих с кровью продуктов их окисления, вступающих во взаимодействие с рецепторами-мусорщиками наружных клеточных мембран макрофагов, то есть повышает вероятность реализации инициирующего момента атеросклероза.
Глава 12 ВОСПАЛЕНИЕ
ОПРЕДЕЛЕНИЕ И БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ
ВОСПАЛЕНИЯ
Воспаление - это системная защитная реакция уничтожения и элиминации всего чужеродного, которая достигает своей биологической цели в основном посредством:
♦ активации системы комплемента;
♦ дегрануляции тучных клеток;
♦ роста проницаемости микрососудов и адгезивной способности эн
дотелия;
♦ миграции плазмы крови в межклеточные пространства;
♦ адгезии к эндотелиальным клеткам нейтрофилов, моноцитов и лимфоцитов циркулирующей крови и их выхода в интерстиций;
♦ фагоцитоза, бактерицидного и цитолитического действия фагоцитов;
♦ расширения, спазма и тромбоза микрососудов;
♦ замещения дефекта тканей через ангиогенез и пролиферацию фиб- робластов.
Под чужеродным в данном контексте следует понимать не только микроорганизмы и инородные частицы, попавшие во внутреннюю среду, но и свои переродившиеся или некробиотически измененные клетки. Подвергшиеся цитолизу клетки приобретают свойство антигенной стимуляции системы иммунитета, высвобождая антигены для взаимодействия с системой иммунитета, в том числе и фрагменты клеточных мембран. Изменение строения наружной клеточной мембраны некробиотически измененных клеток обуславливает активацию на них системы комплемента по альтернативному пути. Активация системы комплемента вызывает воспаление. Переродившиеся малигнизированные клетки при взаимодействии с системой иммунитета вызывают ее антигенную стимуляцию. При этом на их клеточной поверхности появляются антигены, к которым нет иммунологической толерантности. При извращении реакций иммуной системы нормальные антигены клеточных поверхностей приобретают свойство стимулировать систему иммунитета собственного организма. В результате на поверхности таких нормальных клеток образуются иммунные комплексы, что через активацию системы комплемента по классическому пути индуцирует воспаление. В данном случае клетки не перерождаются, но через потерю иммунной толерантности организма к их поверхностным антигенам приобретают свойство чужеродности.
В этой связи можно выделить следующие основные причины воспаления:
|
|
|
